



CHD is one of the most common birth defects in newborn babies. Reference Lowry1 Although extremely rare, these malformations can lead to sudden cardiac death during early childhood. Sudden cardiac death is a tragic and calamitous event defined as natural death, which is cardiovascular in nature and illustrated through an abrupt loss of consciousness within an hour of the onset of cardiac symptoms. Reference Zeigler and Payne2 The annual paediatric incidence rate of sudden cardiac death can vary depending on age-specific groups but overall, it is estimated to be around 1.7 per 100,000 population and 7.5 per 100,000 children. Reference Chugh, Reinier and Balaji3 It is suspected that 5–10% of sudden cardiac death in these patients is due to CHD and genetically determined primary electrical disturbances. Reference Zeigler and Payne2 The intention of this review is to comprehensively study this interrelation and determine which interventions are efficacious in reducing the incidence of childhood sudden cardiac death in patients with CHD.
Aetiology
There can be multiple causes associated with sudden cardiac death in the paediatric population (Fig 1) Reference Meyer, Stubbs and Fahrenbruch4 and even more underlying causes for the development of CHDs. The asymptomatic nature of many CHDs can contribute to children not being screened appropriately or having relevant cardiac examinations, and often this results in late discovery of the underlying pathology at autopsy.

Figure 1. Aetiology of SCD by age group. Reference Meyer, Stubbs and Fahrenbruch4
Virally induced mechanisms
CHDs can be linked to external risk factors including foetal exposure to retinoic acid as well as viral infections in the mother. Some studies suggest that maternal infection with rubella and cytomegalovirus poses an increased risk the development CHD in the foetus. A recent meta-analysis of observational studies of 67,233 women, showed that mothers who suffered from viral infections in their first trimester of pregnancy resulted in a higher global risk of their offspring developing CHD (odds ratio [OR], 1.83; 95% CI, 1.58–2.12; p < 0.0001). Specifically, the risk of CHD development was greatly increased in cases where mothers had been infected with the rubella virus (OR, 3.49, 95% CI, 2.39–5.11) and cytomegalovirus (OR, 3.95; 95% CI, 1.87–8.36). Reference Ye, Wang and Yang5 Similarly, another meta-analysis of case–control studies, studying 524 children with CHD against 262 controls, demonstrated that maternal upper respiratory tract infection (influenza virus) in early pregnancy was associated with an increased risk of their offspring developing CHD (OR 3.40; 95% CI: 2.05–5.62 for simple CHD cases and OR 2.39; 95% CI: 1.47–3.88 for complex CHD cases). After a meta-analysis, the adverse impact was still kept significant (OR = 1.47 and 95% CI: 1.28–1.67 for simple CHD; OR = 1.44 and 95% CI: 1.14–1.75 for complex CHD), same findings were also seen with ventricular septal defects. Reference Xia, Zhao and Zhao6
The genetic interplay
Another notable cause of CHD development is genetic factors, specifically some aneuploidies and gene mutations. Trisomy 21 (Down’s syndrome) is the commonest chromosomal abnormality associated with CHD Reference Hartman, Rasmussen and Botto7 with the commonest CHDs being an atrioventricular septal defect, ventricular septal defect, atrial septal defect, patent ductus arteriosus, and Tetralogy of Fallot. Reference Allen, Driscoll, Shaddy and Feltes8 Results from a matched cohort Australian study indicated a higher incidence of cardiovascular endpoints in Down’s syndrome patients than those without Down’s syndrome. Not only was the prevalence of CHD higher in the Down’s syndrome cohort, but also the incidence of coronary events such as myocardial infarction was higher. Reference Sobey, Judkins, Sundararajan, Phan, Drummond and Srikanth9 Likewise, another study looking into 93 Down’s syndrome patients found a 16.3% mortality at a mean age of 19 months of age. Reference Abbag10 Despite the small sample size, it is clear that Down’s syndrome patients experience higher mortality rates due to underlying CHDs. The second genetic cause of CHD is Turner’s syndrome, affecting females, with the karyotype (45, XO). Occurring in ≈1 in 2500 live female births, Reference Nielsen and Wohlert11 common cardiac defects associated here commonly include bicuspid aortic valve and coarctation of the aorta. These, along with systemic hypertension, can lead to aortic dilatation and dissection, Reference Sybert12 hence increasing the risk of sudden cardiac death. This also emphasises the requirement for screening echocardiogram upon diagnosing patients with Turner’s syndrome to assess their cardiac health, identify potential pathology, and arrange early intervention if needed. Noonan’s syndrome, an autosomal dominant condition, also presents with CHD. Caused by missense mutations in the PTPN11 gene, which encodes for the phosphatase SHP2, it results in increased activation of the RAS/extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK) pathway. Common CHDs in patients with Noonan’s syndrome include pulmonary valve stenosis, atrial septal defects, and congenital hypertrophic cardiomyopathy and mitral valve defects. Reference Pierpont, Brueckner and Chung13 Sudden cardiac death is a concern in patients with Noonan’s syndrome, particularly due to congenital hypertrophic cardiomyopathy being a major risk factor; for example, Ramond et al in 2017 reported a fatal case of a patient with Noonan’s syndrome, who had a sudden cardiac death event whilst running. Reference Ramond, Duband and Croisille14
Diabetes and CHD
Furthermore, another risk factor that is linked with foetal development of CHD is maternal diabetes mellitus. In particular, hyperglycaemia is thought to be the primary teratogen. Reference Negrato, Mattar and Gomes15 The time period in which the mother develops diabetes can have different effects on foetal development. Reference Basu and Garg16 Whilst developing diabetes mellitus prior to conception may lead to neural tube, vasculature, and cardiac defects, Reference Zhao and Reece17 developing gestational diabetes mellitus later on in the gestational period may lead to congenital cardiomyopathy. Reference Kc, Shakya and Zhang18 To supplement this, a population-based study on 1583 babies of mothers with type 1 diabetes mellitus, demonstrated an increased prevalence of CHD-born babies to mothers with type 1 diabetes mellitus against the background population (OR = 3.5; CI: 2.7–4.7). Reference Eidem, Stene and Henriksen19
Drugs and their teratogenic properties
Many drugs are contraindicated in pregnancy due to their adverse foetal outcomes. Maternal lithium use, for the treatment of bipolar disorder, has been shown to increase the risk of foetal right ventricular outflow tract obstructions. Reference Patorno, Huybrechts and Bateman20 Ebstein’s anomaly, in particular, is a congenital defect of the tricuspid valve and associated with the use of lithium during pregnancy. Reference Patorno, Huybrechts and Bateman20 The poor outcomes linked with Ebstein’s anomaly have been well documented since the 90s. One study assessed the presentation and outcomes of 50 children with Ebstein’s anomaly. Reference Celermajer, Cullen, Sullivan, Spiegelhalter, Wyse and Deanfield21 18% died in the neonatal period, 30% died at a mean age of 4.5 years due to haemodynamic deterioration, and 10% of the patients died suddenly at various different ages. Use of angiotensin-converting enzyme inhibitors during the first trimester has also been reported to have adverse fetal cardiovascular outcomes. A study by Cooper et al analysed a cohort of 29,507 infants of whom there were 209 infants who had been exposed to angiotensin-converting enzyme inhibitors in the first trimester and 202 infants who had been exposed to other anti-hypertensives. Infants exposed to angiotensin-converting enzyme inhibitors were at increased risks of malformations involving the cardiovascular and central nervous systems when compared to infants who had exposure to other antihypertensives or no drug exposure at all. Reference Cooper, Hernandez-Diaz and Arbogast22 Veroniki et al also identified that the use of certain anti-epileptic drugs during pregnancy increased the risk of cardiac malformations. The drug combinations which were identified included gabapentin monotherapy, carbamazepine plus phenytoin, phenobarbital plus sodium valproate, phenytoin plus sodium valproate, and carbamazepine plus clonazepam. However, newer generation anti-epileptics drugs such as levetiracetam and lamotrigine were deemed safer. Reference Veroniki, Cogo and Rios23
Maternal factors
Maternal cigarette smoking during pregnancy is a significant risk factor for CHDs and may account for 1.4% of all CHDs. Reference Sullivan, Dervan, Reiger, Buddhe and Schwartz24 A population-based study by Sullivan et al explored the relation between CHDs and smoking during the first trimester and found that offspring of smoking mothers were at increased risk of CHD, especially pulmonary valve anomalies, pulmonary artery anomalies, and atrial septal defects. Reference Sullivan, Dervan, Reiger, Buddhe and Schwartz24 Another interesting risk factor for the development of CHDs is the use of assisted reproductive technology such as in vitro fertilisation and intracytoplasmic sperm injection. A study in the USA analysed this association and found that conception by assisted reproductive technology had a greater risk of birth defects when compared to spontaneous conception. More importantly, the birth defects most commonly associated were cyanotic heart defects, with a frequency of 0.50% in the assisted reproductive technology group and 0.07% in the spontaneous pregnancy group. Reference Shechter-Maor, Czuzoj-Shulman, Spence and Abenhaim25 This is supported by another study by Shamshirsaz et al who also discovered that fertility treatments led to increased risks in the development of foetal cyanotic heart defects. Reference Shamshirsaz, Bateni and Sangi-Haghpeykar26 It is thought that along with the process of the treatment itself, the parental subfertile substrate may also be an underlying mechanism, and hence the use of assisted reproductive technology warrants screening via fetal echocardiography. Reference Shamshirsaz, Bateni and Sangi-Haghpeykar26
Classification
Congenital abnormalities which lead to sudden cardiac death in children can be classified into congenital structural heart defects, congenital coronary artery anomalies, and congenital cardiac arrhythmias, otherwise known as primary electrical disturbances. Reference Wren27 Table 1 lists the conditions which fall into each category with congenital structural heart defects further subdivided into simple and complex defects. Reference Serinelli, Arunkumar and White28
Table 1. A summary of the congenital cardiovascular defects affecting the paediatric population
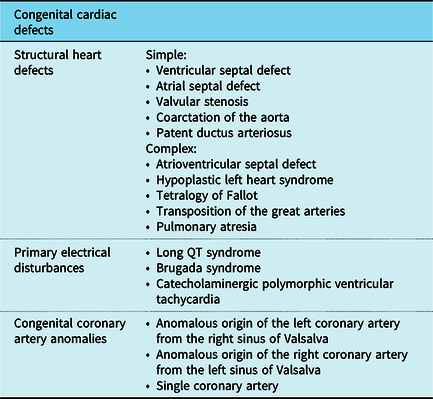
It is important to differentiate between simple and complex structural heart defects as sudden cardiac death in children aged <1 year is more frequently associated with the latter. Reference Serinelli, Arunkumar and White28 One retrospective study identified 64 cases of children with CHDs presenting with sudden death with half of these cases undiagnosed preceding autopsy. 81% of this cohort was infants and the commonest malformation was found to be atrioventricular septal defects, a complex defect. Reference Serinelli, Arunkumar and White28 Complex cardiac congenital defects progressing to sudden cardiac death is further supported in a review by Liberthson which identified literature in which patients with Tetralogy of Fallot had a 6% risk of sudden cardiac death, between 3 months and 20 years of age, after reparative surgery and individuals following an atrial switch operation for transposition of the great arteries were associated with a 2–8% risk of sudden cardiac death. Reference Serinelli, Arunkumar and White28,Reference Liberthson29 Although these findings do not identify the risks associated exclusively with children <18 years, it is still noteworthy as the combination of these findings highlight that the fatal consequences of CHD still linger even after corrective interventions. This places emphasis on the importance of continued perusal in this area of study.
Complex CHDs also tend to coexist with congenital coronary artery anomalies. Reference Lowry, Olabiyi, Adachi, Moodie and Knudson30 Although most cases of congenital coronary artery anomalies tend to be benign, in isolation, congenital coronary artery anomalies can be associated with myocardial ischaemia, ventricular arrhythmias, and sudden death. Reference Frommelt31 Anomalous aortic origin of the left coronary artery from the right sinus of Valsalva accounts for roughly 40% of all major coronary anomalies, however, is deemed to have little clinical significance. Anomalous aortic origin of the right coronary artery from the left sinus of Valsalva is estimated to account for 35% of major coronary anomalies and is important in that it can lead to myocardial ischaemia and sudden cardiac death. Another variant is when there is a single coronary artery and half of the cases are usually encountered alongside other forms of CHD. Reference Flores, Moore and Statile32 Variants with coronary arteries which follow an interarterial course between the aorta and pulmonary artery are thought to have the highest risks of sudden cardiac death, especially when the young individual is engaging with strenuous physical exertion. Reference Brothers33 One study analysed clinical and pathological reports in 242 patients with isolated congenital coronary artery anomalies to identify the frequency and mechanisms pinned with sudden cardiac death. Reference Taylor, Rogan and Virmani34 A key finding was that the younger patients (<30 years old) were at a greater risk of sudden cardiac death, especially following exercise or significant exertion. Although this included patients beyond the paediatric population, it warrants further probing to determine which age groups are most at risk.
Neonates and infants
Congenital malformations are the chief cause of neonatal death, occurring in 2–3% of live births. Structural heart disease is diagnosed in the first year of life in 4–8/1000 infants and 10% of these cases have fatal consequences. The predominant diagnoses made in neonates include hypoplastic left heart syndrome, pulmonary atresia and transposition of the great arteries, all of which are severe and can lead to cyanosis, congestive heart failure, cardiovascular collapse, and shock. Reference Pinar35 In addition, aortic valve stenosis, coarctation of the aorta, and Tetralogy of Fallot are also severe diagnoses, which may be recognised in the first year of life. Sudden unexpected neonatal death and sudden infant death syndrome are both phenomena, which share certain features and have also been shown to have underlying cardiac causes. Reference Weber, Ashworth, Risdon, Brooke, Malone and Sebire36,Reference Adams, Good and Defranco37 A retrospective analysis by Weber et al found that out of 32 explained sudden unexpected neonatal death associated with undiagnosed congenital abnormalities, 12 (38%) were attributable to clinically undetected structural CHD including transposition of the great arteries, coarctation of the aorta, hypoplastic left heart syndrome, and pulmonary atresia. Thus, CHD was the commonest single group of congenital malformations associated with sudden unexpected neonatal death. Reference Weber, Ashworth, Risdon, Brooke, Malone and Sebire36 Although this is a significant value when discussed alongside the sample size and should be included when assessing the burden of sudden cardiac death in paediatric CHD patients, it is believed that sudden cardiac death is more likely to occur in patients beyond the neonatal period and a third of CHD deaths occur during infancy.
Sudden infant death syndrome is most commonly related to environmental factors such as sleeping positions, sleeping environment, and smoking during pregnancy. However, cardiac disorders and especially congenital arrhythmias are responsible for a small number of cases. Reference Adams, Good and Defranco37 Congenital arrhythmias encompass a spectrum of different electrical disturbances including Brugada syndrome, long QT syndrome, and catecholaminergic polymorphic ventricular tachycardia, amongst others. In particular, the long QT syndrome has been suggested to have strong associations with sudden cardiac death in early childhood. Reference Saul, Schwartz, Ackerman and Triedman38 It is commonly known to progress towards torsades de pointes, ventricular tachycardia and fibrillation manifesting as seizures and syncope in seemingly healthy individuals, and so it is understandable as to why this link has been speculated in the past. One genetic study by Arnestad et al demonstrated that 9.5% of patients who suffered from sudden infant death syndrome were found to carry functionally significant variants of long QT syndrome genes, which may have contributed to this premature death. However, without electrocardiogram evidence prior to death, it is difficult to conclude a definite relation between long QT syndrome and sudden infant death syndrome. Reference Arnestad, Crotti and Rognum39 Another genetic analysis in Korea also identified a number of sudden infant death syndrome victims (7.5%) who carried gene variants associated with long QT syndrome, but then found that this variation may also be a common polymorphism in Asians and hence failed to demonstrate a favourable link between long QT syndrome and sudden infant death syndrome. Reference Son, Kim, Yang, Choi, Lee and Yoo40 A key limitation of both of these studies is that DNA investigations use anonymised data to draw conclusions, and hence, this prevents identification and provision of prophylactic therapy to relatives of the deceased individuals who may also be mutation carriers and at increased risk of sudden cardiac death. However, if the overall results fail to show any meaningful relation between long QT syndrome and sudden infant death syndrome then prophylaxis, in any case, may not be beneficial. The results from both of these studies show that whilst some genetic links have been identified, further objective assessments and studies are needed in order to support previous claims of sudden death during childhood in patients with long QT syndrome. Reference Saul, Schwartz, Ackerman and Triedman38
Screening and prevention
A crucial element in improving mortality rates from sudden cardiac death is prevention. In such cases, earlier the patient is diagnosed and treated, the better survival chances. However, apart from primary prevention, secondary prevention strategies have been implemented with the aim of deterring further cardiac damage. Screening is the first step forward. With sudden cardiac death occurring in young individuals in seemingly good health, screening for potential risk factors is essential, particularly when these individuals lead an athletic lifestyle. Reference Maron, Doerer, Haas, Tierney and Mueller41 Exercise exacerbates the effects of CHD, however, 73% of sudden cardiac arrest has been reported to occur when the patient is sedentary. Reference Allan, Morrison, Pinter, Tu and Dorian42 In a study by Mahoney et al 1996, 13% of individuals with sudden cardiac arrest between 2 and 24 years were found to have at least one cardiovascular risk factor. Reference Mahoney, Burns and Stanford43 For this reason, dietary and safe exercise has been publicly recommended. Genetic analysis of sudden cardiac arrest in patients can still be beneficial for relatives. Cascade testing is an important screening mechanism to identify causative mutations, considering a likelihood of 25–50% of first-degree relatives experiencing sudden cardiac arrest in their lifetime. In a retrospective study at a tertiary paediatric referral centre, following evaluation of paediatric patients with a family history of sudden death, 9.3% were diagnosed with a hereditary cardiac disease with Q T syndrome being the most common diagnosis. In 5.5% of all patients, non-hereditary cardiac disease diagnosis had been made and in another 12.4%, findings of uncertain significance resulted. Reference Webster, Olson and Schoppen44
For selecting patients with likely sudden cardiac arrest, algorithms should be developed pivoting around symptoms and their manifestation in clinical events. The risk of sudden cardiac arrest in individuals with a history of aborted or resuscitated sudden cardiac arrest is high, other symptoms that may aim in diagnosis include palpitations, chest pain, syncope, and seizures all generally linked to non-sustained arrhythmia. Between 25 and 50% of children with sudden cardiac arrest have experienced previous symptoms, normally syncope and seizure. Reference Winkel, Risgaard, Sadjadieh, Bundgaard, Haunso and Tfelt-Hansen45 Notwithstanding, apart from syncope, such antecedent symptoms are very common in children and adolescents introducing imprecision in prediction. Additionally, screening strategies have been suggested by the National Heart Lung and Blood Institute. Due to cost–benefit reasons, cardiovascular screening should focus on high-risk groups, such as athletes since asymptomatic screening of young individuals has proved to be ineffective. A multitude of assessment methods have been suggested with ECG, physical examination, and history taking as the best contenders. Reference Bagnall, Singer and Tfelt-Hansen46 Whilst algorithms may enhance the efficiency of ECG delivery, the precision and accuracy of these tests are dependent on population prevalence, age, and medication in use. Risk stratification for paediatric patients may also be limited to specific disease criteria, for example, diagnosis of hypertrophic cardiomyopathy and channelopathies. However, being based on adult guidelines, their validity for paediatric population may be contested. Reference Priori, Wilde and Horie47
Following identification of high-risk individuals, suitable precautionary measures should be taken. An example of this is implantable cardioverter-defibrillator implantation. Findings on the application for implantable cardioverter-defibrillator in paediatric patients are conflicting. Evidence on their use for primary prophylaxis in children is less substantial than in adult counterparts. The risk in implantation of implantable cardioverter-defibrillator is, however, higher in paediatric patients due to lead malfunctions, shock frequency, and difficulty experienced in implantation resulting from the small size of patients and intracranial structures. Reference DeWitt, Triedman and Cecchin48
Another key feature of secondary prevention is cardiopulmonary resuscitation and automated external defibrillators. In an American study, emergency medical service staff were trained to make a preliminary assessment of the cause of sudden cardiac arrest before initiating a cause-specific cardiopulmonary resuscitation. This resulted in better outcomes than in cases when the sudden cardiac arrest cause was not identified to be cardiac or non-cardiac. However, due to time limitations, the extent of pre-cardiopulmonary resuscitation assessment may be restricted. 49 Cardiopulmonary resuscitation training programmes for family members and teachers of paediatric patients with CHD Reference Blewer, Putt and Becker50 together with the provision of self-instruction kits is another facet of secondary prevention aimed at increasing the survival rate from sudden cardiac arrest. Reference Pierick, Van Waning, Patel and Atkins51 Nonetheless, results from a multicentre study suggest that cardiopulmonary resuscitation alone is not as effective as cardiopulmonary resuscitation in conjunction with automated external defibrillators availability. With out-of-hospital cardiac arrest outcomes being low and survival rate being reduced by 10% for each minute cardiopulmonary resuscitation is delayed, Reference Cummins, Ornato, Thics and Pepe52 cardiopulmonary resuscitation-automated external defibrillators programmes have become increasingly popular in community settings. This stems from the fact that some individuals may have not been identified via screening and cardiac arrest is the first presentation of their underlying heart disease. Reference Maron53 Such programmes have been recommended through guidelines by the American Heart Association, indicating that schools with high-risk individuals for sudden cardiac arrest such as patients of cardiomyopathies, channelopathies, congenital, or known cardiovascular disease would require the onsite presence of automated external defibrillators machines. 54 With automated external defibrillators still being inaccessible to some areas, wearable off-brand automated external defibrillators might also provide a solution. Reference Everitt and Saarel55
Discussion
Overall, this review has canvassed a number of causes of CHD encompassing viral, genetic, endocrinological, drug related, and maternal. The prevalence of these aetiologies has been discussed and studies of statistical significance have been pulled apart to closely analyse the increased risk of developing CHDs in these individuals. With regards to viral aetiology, maternal infection with rubella and the influenza virus were the most closely related to CHD. The meta-analyses which highlighted these results showed no publication bias and hence this is indicative of influential credibility. The large sample sizes selected in these studies further display a lack of bias. Furthermore, we were able to identify which genetic conditions had the highest risk of cardiovascular complications. Down’s syndrome, Turner’s syndrome, and Noonan’s syndrome are widely known to have severe cardiac manifestations and unfortunately in these patients, sudden death can occur during childhood owing to severe instabilities of the circulatory system. We reviewed case reports of children to identify those with genetic disorders suffering from sudden cardiac death, and these were based on individual cases or a few patients at a time and therefore, unfortunately, our conclusions cannot be generalised to include different epidemiological populations. There are a vast number of causes for CHD which exist in different proportions according to geographic locations and so patient demographics are a significant varying factor. The most important drugs identified which were shown to be directly linked to birth defects, especially cardiovascular, were lithium, angiotensin-converting enzyme inhibitors, and a number of anti-epileptic drugs.
CHD and structural heart disease form one part of a spectrum of congenital cardiac disorders, which predispose an individual to sudden cardiac death, and so we found that further classification of disease types based on anatomy and physiology aids in the understanding of the specific mechanisms underlying premature death. As a result, we were able to include our assessment of not only structural heart defects, but also congenital coronary artery anomalies and primary electrical disturbances. This review found that those CHDs considered being complex in both pathophysiology and management were more likely to lead to sudden cardiac death. The small sample sizes discussed, however, may negatively impact our deductions and this mandates further studies analysing these conclusions. Complex CHDs sometimes also appear alongside congenital coronary artery anomalies and these in isolation increase the risk of sudden cardiac death, therefore, it can be hypothesised that the combination of both can have dire consequences. Having reviewed the literature available, it is our opinion that there is insufficient research exposing the links between sudden unexpected neonatal death and sudden infant death syndrome with CHD, and therefore this link should have an important role in future research. Long QT syndrome has been hypothesised to have a role in sudden cardiac death in children <1 year. We managed to explore this through genetic studies; however, both demonstrated a weak or insignificant relationship. Electrocardiogram evidence of pathology would be useful to document in future studies in order to uncover the exact nature and causes of death.
A key part of our aim was to highlight strategies, which could reduce mortality rates in children with CHD. Our thorough analysis of the primary and secondary prevention strategies, which have been implemented so far has allowed us to understand the pivotal role of screening in those with cardiovascular risk factors and the importance of widespread cardiopulmonary resuscitation training and availability of automated external defibrillators. Implantable cardioverter-defibrillator implantation in those children at risk is debatable and this is a decision, which should be made on a case-by-case basis through the involvement of senior clinicians and via a multidisciplinary approach. The literature reviewed has allowed us to appreciate that defibrillation outcome for children and adolescents with sudden cardiac arrest is poorer than that in adults. The discrepancy for this is still elusive. Moreover, the safety of automated external defibrillators for infants younger than 1 year has not been safely established. Instead of direct application of knowledge about adults with CHD, more research about the suitability of screening as well as implantable cardioverter-defibrillator implantation should be conducted specifically targeted towards a paediatric population.
Conclusion
The high incidence of sudden cardiac death in paediatric populations, in particular in younger patients has made it a pressing issue for clinicians and whilst a plethora of factors may contribute to sudden cardiac death, CHD is a known precursor. Successful classification along with continued surveillance of the type of CHD displayed may be a crucial determinant in ameliorating survival of patients. With primary and secondary prevention strategies studies underway, more emphasis is required on the difference between adult and paediatric patients. This review attempted to discuss both established frameworks in sudden cardiac death related to CHD as well as identify potential areas of study. For this reason, a more proactive approach should be encouraged in studying the effectiveness of existent measures and for a more profound understanding of the association between sudden cardiac death and CHD.
Acknowledgements
None.
Financial support
This research received no specific grant from any funding agency, commercial, or not-for-profit sectors.
Conflicts of interest
None.



 Open access
Open access