



Focal myositis (FM) is a localized inflammatory pseudotumor characterized by sudden-onset inflammation in one skeletal muscle. Reference Devic, Gallay, Streichenberger and Petiot1,Reference Milani, Mazzoni, Gatti, Bertolozzi and Fossali2 Contrary to other forms of inflammatory myositis, FM is usually not associated with systemic symptoms or muscle weakness, though fever is occasionally reported. Reference Devic, Gallay, Streichenberger and Petiot1 FM is typically isolated to one muscle and resolves spontaneously within weeks without lasting symptoms. Cases of relapsing symptoms have been reported in up to 41% of adult patients, Reference Gallay, Hot, Petiot, Thivolet-Bejui, Maucort-Boulch and Streichenberger3 but relapsing FM remains poorly described in children. Here, we present the first case, to our knowledge, of bilateral migrating relapsing FM in a child.
A 12-year-old male, born in Canada to a Rwandan mother and Haitian father, presented with gradual bilateral neck pain and neck enlargement over the course of 4–10 days. The patient showed no systemic symptoms or muscle weakness. Pregnancy and delivery were uneventful. Psychomotor development was normal. The patient reported three episodes of myalgia in the preceding year – the first in the left thigh, the second in the right foot, and the third in both thighs. The patient consulted in person for the first two episodes; on both occasions, inflammatory markers were slightly elevated, creatinine kinase levels were normal, and magnetic resonance imaging (MRIs) of the affected muscles showed T2 hyperintensity with limited involvement of surrounding connective tissue, consistent with myositis. Reference Gallay, Hot, Petiot, Thivolet-Bejui, Maucort-Boulch and Streichenberger3,Reference Teixeira, Peixoto and Costa4 All episodes resolved within 2 weeks. The patient was otherwise healthy and took no medications at home. Family history was unremarkable.
The patient, who weighed 54.4 kg, was started on Naproxen 500 mg twice daily for 3 weeks. Like his previous episodes, the patient had mildly elevated inflammatory parameters but normal creatinine kinase and aldolase levels. MRI of the sternocleidomastoid muscle (SCM) muscles showed T2 hyperintensity, consistent with his prior visits. His symptoms improved throughout his 6-day hospitalization. Extensive auto-immune and infectious workups (including a comprehensive myositis-specific autoantibodies panel) were negative. Because of the risks and potential morbidity involved in a SCM biopsy, no biopsy was conducted. Given the patient’s rapid clinical improvement, lack of systemic symptoms, and normal ancillary investigations across multiple visits, alternative diagnoses such as vasculitis and inflammatory myositis were excluded. On follow-up, the patient remained asymptomatic 2 weeks after the end of his Naproxen regimen. A detailed clinical and paraclinical description of the patient’s myalgia episodes is provided in Table 1. Figure 1 presents MRI images of the muscle groups involved in all three hospital visits.
Table 1: Detailed clinical and paraclinical description of the patient’s myalgia episodes
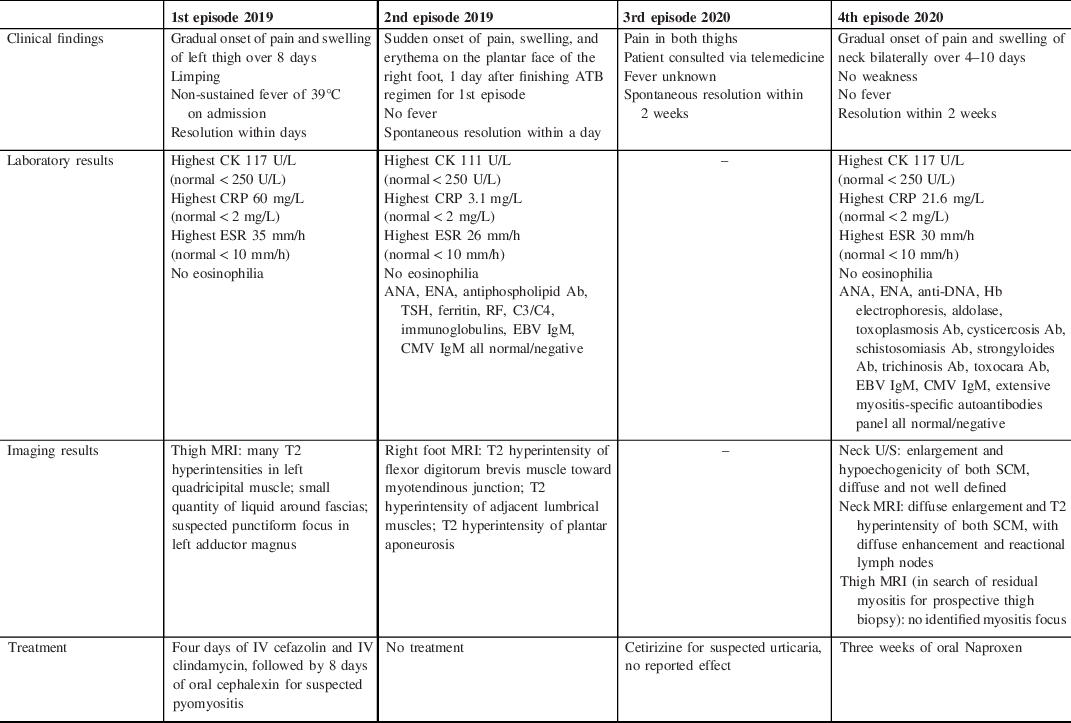
Ab = antibody; ANA = antinuclear antibodies; ATB = antibiotic; CK = creatine kinase; CMV = cytomegalovirus; CRP = C-reactive protein; EBV = Epstein–Barr virus; ENA = extractable nuclear antigen; ESR = erythrocyte sedimentation rate; Hb = hemoglobin; MRI = magnetic resonance imaging; SCM = sternocleidomastoid muscle; TSH = thyroid stimulating hormone.
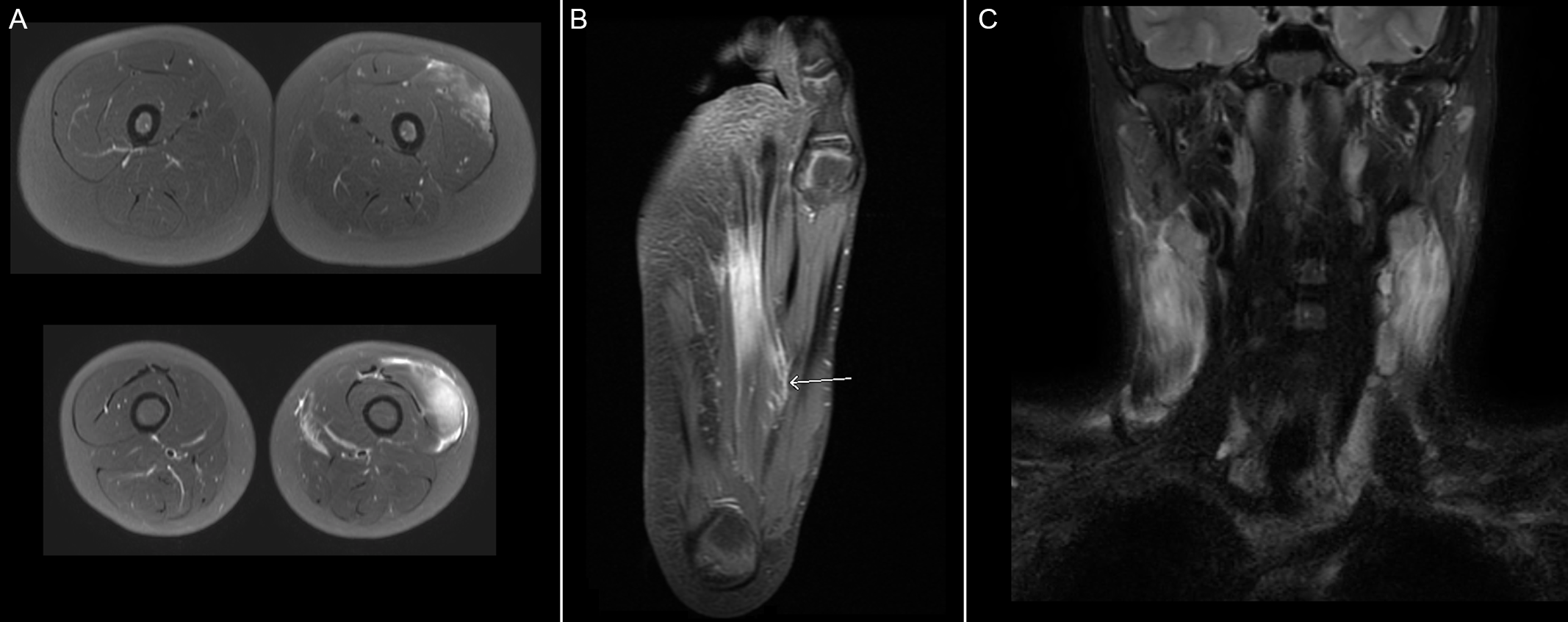
Figure 1: MRI images of muscle groups involved during respective episodes of myalgia. (A) Axial T2 MRI images of thighs during 1st episode of myalgia demonstrating signs of myositis in the left quadricipital muscle. (B) Coronal T2 MRI image of right foot during 2nd episode of myalgia demonstrating signs of myositis in the plantar region of the foot and concomitant plantar fasciitis. (C) Coronal T2 MRI image of the cervical region during 4th episode of myalgia confirming diffuse enlargement, hyperintensity, and enhancement of sternocleidomastoid muscles, compatible with bilateral myositis.
FM is a rare disease that usually presents as self-resolving inflammation of a single muscle, typically the calf, forearm, or thigh. Atypical presentations have been reported, although SCM involvement has only been noted in adults so far. Reference Devic, Gallay, Streichenberger and Petiot1,Reference Milani, Mazzoni, Gatti, Bertolozzi and Fossali2,Reference Bennett, Kumar and Agnew5 Systemic symptoms and muscle weakness are usually absent. The disease affects both sexes equally and is most common in middle-aged adults, though individuals of all ages can be affected. Reference Devic, Gallay, Streichenberger and Petiot1
The disease has no known etiology. Nerve lesions, infectious diseases, neoplasms, and immune disorders – notably Behcet’s disease – are proposed triggers for FM. Reference Devic, Gallay, Streichenberger and Petiot1 Some reported cases of FM evolve into polymyositis (PM) or dermatomyositis (DM), but such an evolution is rare and may represent a misdiagnosis of an early form of PM that manifests as localized nodular myositis. Reference Milani, Mazzoni, Gatti, Bertolozzi and Fossali2,Reference Teixeira, Peixoto and Costa4 The relationship between FM and other inflammatory pseudotumors, such as idiopathic orbital inflammation, has not been studied. Whether these entities represent separate conditions or different manifestations of a common underlying etiology remains unknown.
The disease course is variable, with relapse occurring in up to 41% of adult cases. Reference Gallay, Hot, Petiot, Thivolet-Bejui, Maucort-Boulch and Streichenberger3 Bilateral relapses have only been reported in adults. Reference Nagafuchi, Nakano and Ooka7 Clinical evolution is benign; however, a diagnosis of FM may signal an underlying disease. In a 2018 study, FM was associated with immune-mediated inflammatory diseases (IMIDs) in up to 32% of patients, as well as with neoplasia in 24% of patients and radiculopathy in 11% of patients. Reference Gallay, Hot, Petiot, Thivolet-Bejui, Maucort-Boulch and Streichenberger3 Prompt diagnosis of FM may thus help discover treatable underlying conditions.
FM is a diagnosis of exclusion. Efforts have been made to build a diagnostic score, but no consensus has been reached yet on definite criteria. Reference Gallay, Hot, Petiot, Thivolet-Bejui, Maucort-Boulch and Streichenberger3 Diagnosis relies on a holistic evaluation of clinical presentation, imagery, laboratory results, and pathology. Biopsy can help rule out certain alternative diagnoses; however, since histological analysis is generally non-specific and occasionally presents atypical features, some authors suggest that biopsies only be performed in the presence of systemic symptoms or persistent inflammation despite appropriate treatment. Reference Milani, Mazzoni, Gatti, Bertolozzi and Fossali2,Reference Jun, Im, Park, Yoo and Park6
FM remains poorly studied in children. We report an unusual case, where a 12-year-old child presented with bilateral relapsing migrating FM. His symptoms and negative ancillary investigations were consistent with cases of pediatric FM. Reference Devic, Gallay, Streichenberger and Petiot1,Reference Bennett, Kumar and Agnew5 Moreover, his history of localized episodes with rare systemic symptoms, no muscle weakness, and persistently normal creatine kinase levels would be highly unusual for PM/DM, vasculitis, or a genetic condition such as PAPA or SAPHO. MRI results further supported our diagnosis. Although a biopsy was not performed, the non-specific histological findings of FM would not have provided much diagnostic value in this patient’s case, given the lack of systemic symptoms on multiple visits. Reference Milani, Mazzoni, Gatti, Bertolozzi and Fossali2
Treatment of FM is typically limited to anti-inflammatory drugs. Reference Devic, Gallay, Streichenberger and Petiot1,Reference Milani, Mazzoni, Gatti, Bertolozzi and Fossali2 It is uncertain if steroid regimens should be considered in cases where inflammatory markers remain normal. Some immunosuppressors have been used in the literature, with no consistent outcome. Reference Devic, Gallay, Streichenberger and Petiot1 Long-term follow-up is suggested because of the potential, unconfirmed risk of PM/DM in these patients. Physicians should be careful when diagnosing FM so as not to misdiagnose the early showings of an underlying systemic disorder such as PM/DM or eosinophilic myositis. Migrating FM should be suspected in patients presenting with focal inflammation of one muscle group in the absence of systemic symptoms suggestive of alternative diagnoses.
To our knowledge, this is the first pediatric case of bilateral relapsing migrating FM. This is also the first case of sternocleidomastoid involvement in a child. Given the variety of clinical presentations and the association between FM and conditions such as IMIDs and neoplasia, we believe more research is required to elucidate the physiopathology of FM and its relation to other auto-immune and inflammatory disorders.
Disclosures
The authors have no conflicts of interest to disclose.
Statement of Authorship
LLH: role in data collection and manuscript redaction; JL: role in data collection and manuscript redaction; MPM: role in data collection and manuscript revision; JJB: role in data collection and manuscript revision; BT: role in data collection and manuscript revision; PM: role in data collection and manuscript revision, corresponding author.


