



Immunoglobulin light chain (AL) amyloidosis is a plasma cell disorder leading to monoclonal light chain deposition in various tissues including heart, liver, gastrointestinal tract, kidneys, peripheral nerves, and less commonly muscle. Reference Muchtar, Derudas and Mauermann1 AL amyloid myopathy is a rare presentation of AL amyloidosis and represents roughly 1.5% of all patients with AL amyloidosis according to the largest available study of this patient group. Reference Muchtar, Derudas and Mauermann1 Common features of amyloid myopathy (ALM) include proximal muscle weakness, dysphagia, myalgias, and macroglossia. Creatine kinase (CK) levels can be normal. Reference Muchtar, Derudas and Mauermann1,Reference Liewluck and Milone2 Prognosis is poor given delayed diagnosis and absence of specific therapies. We report two ALM cases with concurrent light chain multiple myeloma (LCMM) that were responsive to plasmapheresis.
Case 1 is a 70-year-old male with kappa LCMM and cardiac and renal amyloidosis. Preceding his LCMM, he had a 10-year history of polyneuropathy with enhancement of his cauda equina and an intrathecal nodule at L3 on magnetic resonance imaging (MRI) (Figure 1). Years prior, an extensive work-up for nutritional, infectious, neoplastic, and inflammatory polyneuropathy was negative. Cerebrospinal fluid (CSF) protein was elevated on two occasions (0.77 and 0.94 g/L), with normal cell counts. Computed tomography (CT) chest, abdomen, and pelvis were normal. Nerve conduction studies (NCS) demonstrated a mild length-dependent sensory axonal polyneuropathy unresponsive to trials of prednisone, intravenous immunoglobulin (IVIg), and methotrexate.
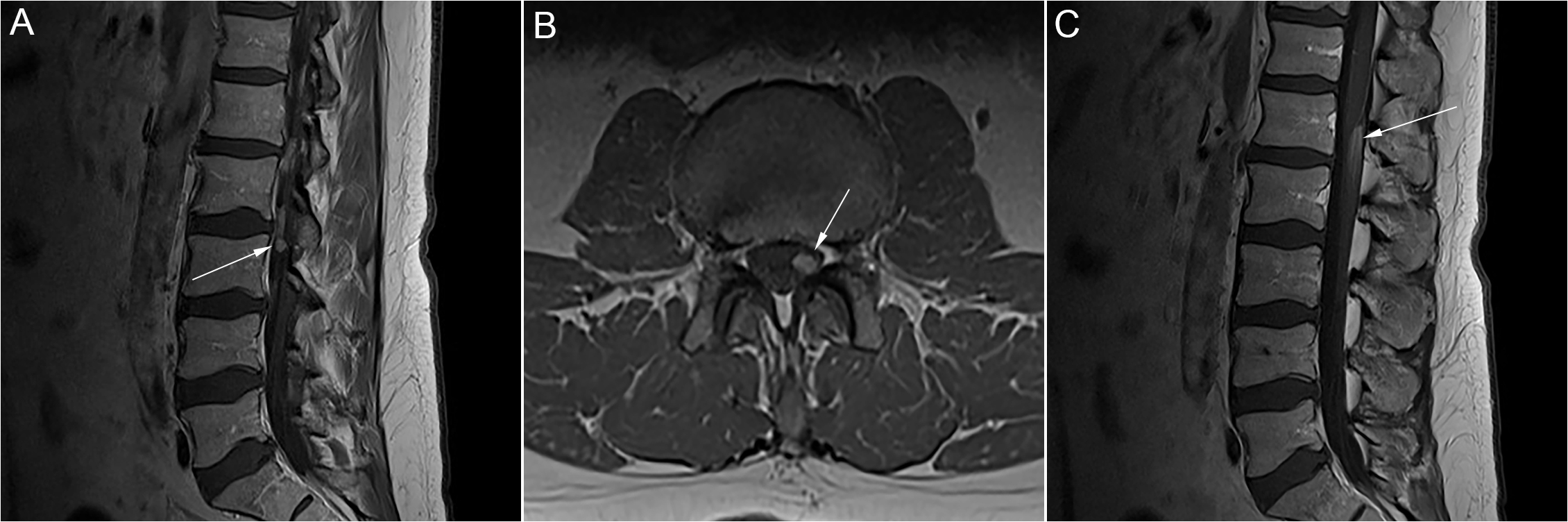
Figure 1: Case 1. A. Sagittal T1 post-gadolinium MRI showing an avidly enhancing 6 mm nodule within the left side of the thecal sac at the level of L3. B. Axial T1 post-gadolinium MRI showing this same nodule. C. Sagittal T1- post-gadolinium MRI showing an ill-defined area of enhancement involving the posterior aspect of the conus and cauda equina at L1/L2, measuring roughly 18 mm in craniocaudal dimension.
One month after diagnosis of LCMM, the patient developed subacute, proximal, and symmetric weakness, which progressed despite CyBorMe (cyclophosphamide, bortezomib, high-dose methylprednisolone) treatment. Treatment was discontinued after four consecutive cycles (four weeks of chemotherapy per cycle) due to his neurologic decline despite his post treatment staging showing normalization of light chains and achievement of stringent complete response (sCR) with minimal residual disease (MRD) detectable by flow cytometry on bone marrow aspirate. Medical Research Council (MRC) grade power was 4− shoulder abduction and elbow flexion, 4+ elbow extension, and 2 hip flexion and knee extension. Distal sensory symptoms worsened after CyBorMe and reflexes were lost. CK and myositis antibodies were normal. NCS/electromyography (EMG) demonstrated an irritable myopathy and no significant motor neuropathy. Right deltoid muscle biopsy showed widespread amyloid deposition (Figure 2). MRI showed progression of cauda equina nerve root enhancement.
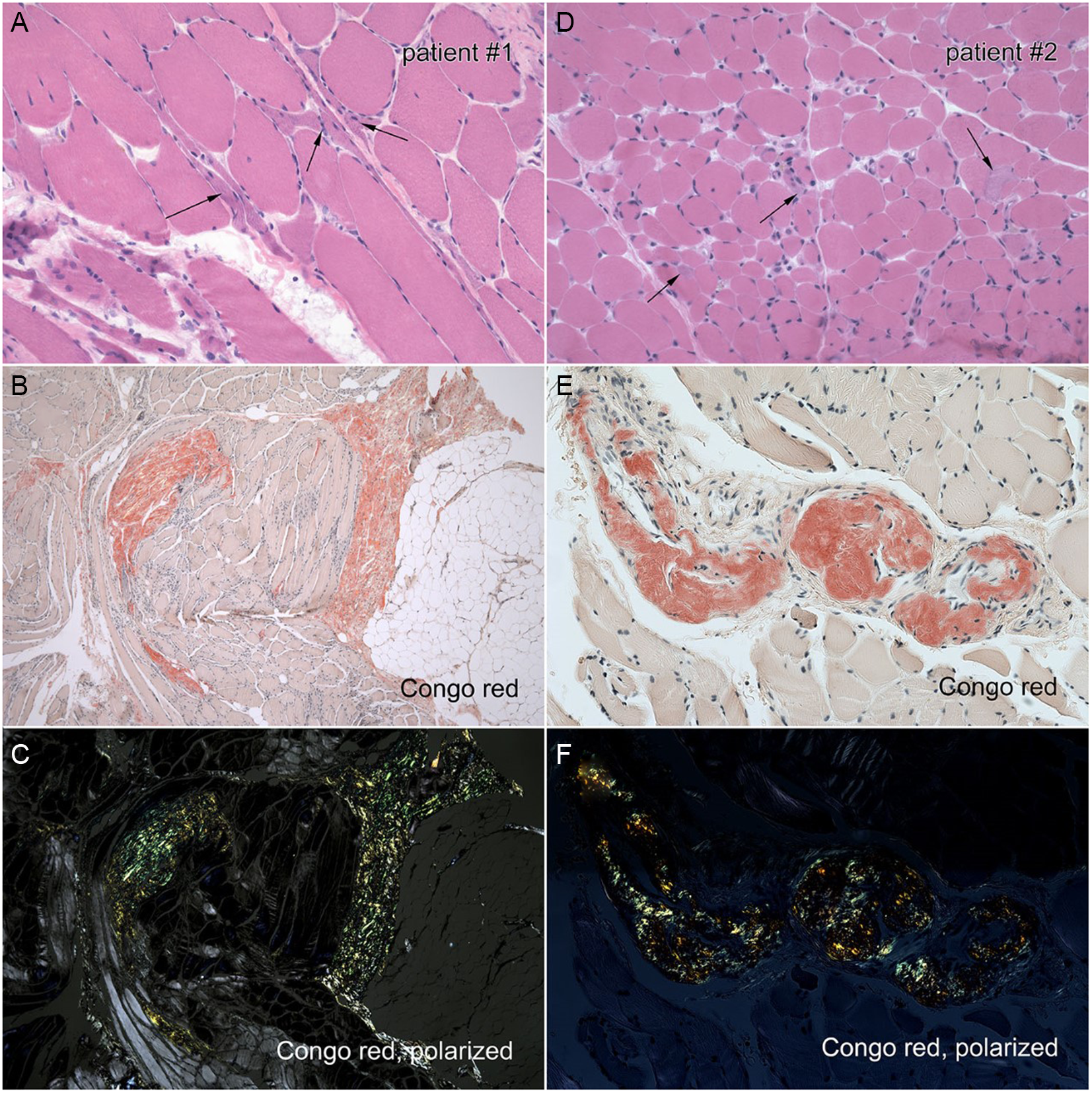
Figure 2: Left panel: Right deltoid biopsy of Case 1. A. H&E stain demonstrating angulated fibers. B. Congo red stain showing endomysial and perimysial amyloidosis C. Congo red, polarized light showing endomysial and perimysial amyloidosis. Right panel: Left biceps biopsy of Case 2. D. H&E stain demonstrates variable fiber size. E. Congo red stain demonstrating perivascular amyloidosis. F. Congo red, polarized light demonstrating perivascular amyloidosis.
Rapidly progressive weakness prompted plasmapheresis therapy at three exchanges per week. After 7 weeks, MRC grade 4 hip flexion was the only remaining weakness. Repeat spine MRI was unchanged. Plasmapheresis was tapered to monthly. At one-year follow-up, strength was normal, reflexes returned in the arms and remained absent in the legs. Sensory exam was unchanged. Repeat right deltoid biopsy showed qualitatively less amyloid, although still abundant. Several months following plasmapheresis discontinuation, motor power remained normal. Sensory exam improved with return of patellar reflexes, negative Romberg, and ability to perform tandem gait.
Case 2 is a 67-year-old female with kappa LCMM, concurrent AL amyloidosis, and proximal weakness. She had multiple vascular comorbidities and right-sided heart failure. A year long history of proximal symmetrical weakness preceded the diagnosis of LCMM, such that she was nonambulatory and required assistance with activities of daily living. She received only three cycles of CyBorMe, due to poor tolerance, however, achieved normalization of light chains and achievement of sCR with MRD positivity by flow cytometry on bone marrow aspirate.
Examination revealed symmetrical MRC graded power of 4 neck flexion, 4− shoulder abduction, 4+ elbow flexion and extension, 2 hip flexion, 4 knee flexion, and 4+ knee extension. Distal power was preserved. Reflexes were absent at the knees only. Pinprick and vibration sensation were intact. EMG demonstrated a nonirritable myopathy. Muscle biopsy demonstrated prominent amyloid vasculopathy (Figure 2). She was treated with plasmapheresis, three exchanges per week for two weeks followed by twice a week for two weeks. Power improved to grade 4+ shoulder abduction, 3 hip flexion, and 4+ knee flexion. Plasmapheresis was continued weekly for 8 weeks and the patient regained the ability to ambulate and climb stairs with a railing. Repeat biopsy showed unchanged amyloid vasculopathy.
ALM outcomes are poor with 32-month median survival, owing to delayed diagnosis and treatment inefficacy. Reference Muchtar, Derudas and Mauermann1 These cases suggest plasmapheresis may be a novel, potentially effective treatment for ALM as an adjunct to traditional clone directed therapy. Previously, plasmapheresis has only shown benefit in systemic amyloidosis with renal complications. Reference Drew4 Current treatments for ALM, including melphalan, daratumumab, and bortezomib-containing regimens, are also used to target the underlying clone but have variable effects in improving ALM. In both cases presented, motor power only improved after plasmapheresis, and improvement was sustained despite discontinuation.
Mechanisms by which plasmapheresis may treat ALM are speculative. Plasmapheresis primarily removes proteins that drive inflammation or deposit in various tissues. Reference Drew4 Although plasmapheresis may limit amyloid deposition in muscle, in both cases, amyloid burden was largely unchanged on repeat biopsy following clinical recovery. Despite this, the degree of amyloid that needs to be removed to result in qualitative changes on pathology is unknown and perhaps smaller changes may be sufficient to drive clinical improvement. It is also possible that there was a delayed benefit from CyBorMe treatment that coincided with plasmapheresis; however, muscle weakness progressed despite clear improvement in LCMM, and recovery was rapid after plasmapheresis initiation.
It is important to consider that weakness in these cases may not have been due to ALM, and plasmapheresis was treating an alternate pathology. In case 1, the cause of cauda equina enhancement is unclear and may suggest an acquired demyelinating neuropathy. However, the patient’s sensory symptoms and nerve root enhancement were present several years prior to weakness developing, demyelinating features were absent on NCS, and there was no improvement in sensory symptoms with IVIg or steroids. Sensory symptom improvement was significantly delayed compared to motor recovery, likely reflecting recovery of toxic neuropathy after chemotherapy. Cauda equina gadolinium enhancement, typically a marker of active demyelinating disease, was unchanged after clinical improvement and remains unexplained. Reference Duggins, McLeod and Pollard5
These cases highlight a potential role for plasmapheresis in ALM treatment. However, the possible mechanism of action remains unclear and further evidence is needed to support the standard use of plasmapheresis. Given poor outcomes in ALM, a plasmapheresis treatment trial may be reasonable in patients with rapidly progressive weakness.
Disclosures
Dr Christopher Hahn, Dr Sameer Chhibber, Dr Tefani Perera, Dr Gordon Jewett, Dr Shahin Khayambashi, and Dr Jeffrey T. Joseph have no conflicts of interest or disclosures. Dr Sylvia McCulloch serves on the advisory board for Sanofi, Jansen, and FORUS. She has received support for attending meetings from Jansen and payment from Pfizer for educational activities, none of which conflict with the contents of this article.
Statement of authorship
Case written and reviewed by TP, SK, GJ, CH, SM, JTJ, and SC. Figures created by GJ and JTJ.



 Open access
Open access