


Case Presentation
This 17-year-old girl was referred to Neurology for assessment of bitemporal headaches in the context of known spastic paraparesis and cognitive dysfunction.
She was born to a healthy 23-year-old mother of Indian origin and father of Pakistan descent, with no family history of neurological disease. Her two siblings were healthy. Her perinatal history was unremarkable. Up until the age of 3, she achieved her developmental milestones. At 3 years old, she was noticed to have in-turning of both feet. Gait abnormalities progressed such that by age 7, she had presented multiple times to the emergency department for falls and related head injuries. She also had associated significant leg pain. At this point, she was given the diagnosis of cerebral palsy (CP), spastic diplegia subtype.
However, by age 10, it was noted that the spasticity was progressive. Her examination was noted as follows: visual acuity was 20/40 bilaterally, with the remainder of the cranial nerve examination being normal. Tone and power in the upper limbs were normal. Upon lower limb examination, muscle bulk was normal. However, tone was spastic with bilateral ankle contractures. Both feet were in-turned. Reflexes were within normal limits in the upper limbs, however, she was hyperreflexic (3+) with ankle clonus in her lower extremities.
By age 15, visual acuity had further decreased to 20/200 bilaterally. An ophthalmologic examination revealed bilateral marked optic nerve pallor. Visual evoked potentials confirmed optic nerve atrophy.
Concerns about cognitive deterioration were raised by her parents and teachers. She repeated grade 2 and grade 9 and was in an individualized education program throughout her schooling.
At age 16, MRI of the brain and spine was performed and reported as normal. Given her progressive spasticity of a complex subtype, a hereditary spastic paraplegia (HSP) panel was sent and this was found to be negative.
By age 18, the patient continued to decline both motorically and cognitively, with increasing spasticity and pain, now requiring walking aids (cane and walker) and leg braces. She was beginning to have swallowing difficulties as well as urge urinary incontinence. No bowel incontinence was reported. She was formally diagnosed with intellectual disability with ongoing cognitive decline. Furthermore, her audiology testing revealed sensorineural hearing loss.
Repeat MRI was subsequently ordered and showed features consistent with mitochondrial membrane protein-associated neurodegeneration (MPAN) (Figure 1). Genetic mutation sequence analysis showed a novel homozygous mutation (c.400G>C) of the c19orf12 gene, consistent with this diagnosis.
Discussion: Spastic Paraplegia
Given the patient’s initial predominant symptom of spastic diplegia, the early presumptive diagnosis was originally CP. CP is the most common cause of pediatric physical impairment and is defined as a group of non-progressive motor disorders due to an insult in the fetal or infant brain. The key in differentiating CP from its mimickers lies in identifying red flags not consistent with CP such as a normal MRI, a normal perinatal history, and neurological and cognitive regression.Reference Huntsman, Lemire, Norton, Dzus, Blakley and Hasal 1 Therefore, it is important to consider genetic conditions and inborn errors of metabolism in this scenario.
The possibility of this patient having HSP was subsequently raised. HSP involves a large group of inherited neurodegenerative corticospinal tract disorders that often present with progressive spasticity, hyperreflexia and lower limb weakness. HSPs can be divided into pure and complex forms: pure HSP only has corticospinal tract degeneration causing motor symptoms whereas complex HSP has additional neurological and extraneurological findings.Reference Cervantes-Ibáñez, Samaranch, Pastor, Kompoliti and Verhagen 2 Mutations in 67 genes have been described and include autosomal dominant (AD), autosomal recessive (AR), and X-linked patterns of inheritance.Reference Tesson, Koht and Stevanin 3
Most cases of HSP are AD (70%) of which the SPG4 mutation is the most common (50% of all HSP). SPG4 is a pure HSP with a variable age of onset.Reference Orsucci, Petrucci and Caldarazzo Ienco 4 More notably, they tend to have an unremarkable MRI.Reference Rezende, De Albuquerque and Lamas 5 SPG11 is the most common of the AR mutations tending to present in childhood and early teenage years featuring gait issues and spasticity. It is considered a complex HSP as it can present with ataxia, parkinsonism, severe bladder problems and cognitive decline. Unlike SPG4, the MRI for SPG11 HSP shows progressive thinning of the corpus callosum and cerebral atrophy following the declining clinical course.Reference Kara, Tucci and Manzoni 6 The clinical course, age of onset, and MRI findings are similar to the findings seen with SPG15, another complex HSP.Reference Pensato, Castellotti and Gellera 7
SPG7 is yet another AR form of HSP presenting with late-onset progressive spasticity and known to have complex variants associated with optic atrophy and ophthalmoplegia. Optic neuropathy, cerebellar findings in conjunction with cerebellar atrophy on MRI are common additional features.Reference Schüle and Schöls 8 , Reference Klebe, Depienne and Gerber 9 The optic nerve involvement together with the similarity in symptoms make it an interesting consideration for the case provided, however, the late age of onset and distinct MRI findings did not fit. Similar findings have also been described for SPG35 with various MRI elements including cerebellar atrophy, corpus callosum atrophy, and white matter abnormalities.Reference Pensato, Castellotti and Gellera 7
Discussion: Neurodegeneration with Brain Iron Accumulation Disorders
While the initial MRI of the patient was not flagged as abnormal, the repeat MRI showed signs suggestive of an iron accumulation disorder. Iron metabolism has been known to be essential to many functions of the central nervous system including but not limited to oxygen transport, DNA synthesis, myelination, mitochondrial respiration, and neurotransmitter regulation.Reference Hare, Ayton, Bush and Lei 10 Gradual iron accumulation occurs naturally with age, seen primarily in the basal ganglia globus pallidus, substantia nigra pars reticulata and putamen as well as the red and dentate nucleus.Reference Morris, Candy, Oakley, Bloxham and Edwardson 11 Many adult-onset neurodegenerative disorders such as Alzheimer’s and Parkinson’s disease have been associated with iron accumulation.Reference Kell 12
The neurodegeneration with brain iron accumulation (NBIA) disorders represent a rare heterogeneous group of neurodegenerative diseases associated with iron accumulation in the basal ganglia. The NBIAs present clinically with progressive pyramidal and extrapyramidal symptoms, neuropsychiatric symptoms, cognitive regression among others.Reference Schneider, Dusek, Hardy, Westenberger, Jankovic and Bhatia 13 , Reference Schneider 14 Among the childhood onset NBIAs, the genetic defects do not involve iron metabolism directly. Instead, the genes are part of various pathways involving coenzyme A production, phospholipid membrane modeling and stability, autophagy, and oxidative respiration.Reference Kurian, McNeill, Lin and Maher 15
A significant game-changer in the identification of NBIAs was the introduction of MR imaging, which now plays a large role in differentiating amongst the various forms. T2-weighted (T2W), gradient echo and susceptibility-weighted imaging (SWI) are especially sensitive to iron deposition and are effective in highlighting characteristic NBIA imaging patterns.
Pantothenate kinase-associated neurodegeneration (PKAN) is the most common NBIA disorder classically presenting in early childhood with both pyramidal (spasticity, hyperreflexia) and extrapyramidal components and often associated with orolingual-mandibular involvement.Reference Hayflick 16 It represents a classic example with its hallmark MRI “eye of the tiger” sign comprising of a hypointense globus pallidus with prominent central anteromedial hyperintensity on T2W imaging.Reference Kruer, Boddaert and Schneider 17 The pathology correlates well with the imaging demonstrating high levels of iron concentrated within the globus pallidus interna.Reference Kruer, Hiken and Gregory 18
The second most common NBIA syndrome PLA2G6-associated neurodegeneration (PLAN) has an early onset form known as infantile neuroaxonal dystrophy (INAD), which involves profound hypotonia, peripheral neuropathy, cognitive regression along with cerebellar ataxia, and optic atrophy.Reference Morgan, Westaway and Morton 19 Interestingly enough, these patients tend to show vermian-cerebellar atrophy, claval hypertrophy, and a vertically positioned corpus callosum early on that precedes the iron accumulation in the globus pallidus. The iron accumulation in PLAN appears quite homogeneous in the GP.Reference Illingworth, Meyer and Chong 20
Beta propeller protein-associated neurodegeneration (BPAN) is the only X-linked dominant NBIA, described as having a “Rett-like” phenotype and presents with global developmental delay, including speech delay, followed by young adult-onset dystonia, parkinsonism and cognitive decline. The majority of patients are girls, with a few boys with early onset epileptic encephalopathy now being identified. The key finding on MRI involves a hyperintense “halo ring” surrounding a thin-linear hypointense region in the substantia nigra on T1-weighted imaging. Additionally, in BPAN, the substantia nigra appears to have more iron accumulation than the globus pallidus.Reference Hayflick, Kruer and Gregory 21
Fatty acid hydroxylase-associated neurodegeneration (FAHN) is an NBIA presenting with childhood onset spasticity followed by dysarthria, ataxia, and motor regression. This condition has many similarities with PLAN but appears to have a slower rate of degeneration. On MRI, FAHN distinguishes itself with white matter abnormalities and brainstem atrophy and appears to have a later onset iron accumulation in the basal ganglia.Reference Kruer, Paisán-Ruiz and Boddaert 22
Case Discussion: MPAN
The patient had a repeat MRI done at age 17 that revealed marked bilateral hypointensity of the globus pallidi, with a hyperintense streak along the medial medullary lamina, and substantia nigra (Figure 1). SWI confirmed localized iron deposition. In retrospect, the original MRI done at age 13 demonstrated T2 hypointensities in the globus pallidi and substantia nigra which would have been easily identified if SWI had been done at that time. Subsequent genetic analysis demonstrated a missense mutation in the c19orf12 (c.400G>C; p. Ala134Pro) gene. This is the same gene that was originally found to be deleted in 24 patients of a Polish cohort in Hartig et al.’s study in 2011 resulting in a neurodegenerative condition that was later labeled as MPAN.Reference Hartig, Iuso and Haack 23
In this study, the 17kb long c19orf12 gene affected was found to code for two 141 and 152 amino acid protein residues derived from isoforms with transmembrane domains highly conserved in evolution and, similarly to other NBIA implicated genes, shown to localize primarily to mitochondria. Its inheritance is of an AR pattern and both missense and nonsense mutations have been described in various studies.Reference Hartig, Prokisch, Meitinger and Klopstock 24
MPAN has since been described in more than 100 Polish patients,Reference Dušek, Školoudík, Roth and Dušek 25 several Mali patientsReference Landoure, Zhu and Lourenco 26 as well as in a Turkish childReference Kasapkara, Tumer and Gregory 27 and likely makes up a significant portion of the NBIA population. Clinically, MPAN has many similarities with the other NBIA syndromes and patients are characterized as having a childhood onset dysarthria and slowly progressive gait disorder followed by extrapyramidal signs in association with cognitive impairment. In contrast to other NBIAs such as PKAN, the onset is later with a milder course but with more prominent neuropsychiatric and neurocognitive dysfunction. Patients have also been found to have optic atrophy (as found with INAD and FAHN but not PKAN) similar to our patient. Furthermore, MPAN has shown to have the unique feature of motor axonal neuropathy, an element lacking in our patient at this point as she had normal nerve conduction studies.
Radiographically, MPAN shows characteristic iron accumulation in the basal ganglia but, unlike INAD, no cerebellar atrophy and classically, unlike PKAN, does not have an eye-of-the-tiger sign. What appears to be more consistent across various cohorts and within our patient is the presence of hyperintense streaking of the medial medullary lamina between the globus pallida interna and externa on T2 images. This is a sign which has been described as taking time to develop and may have explained the difficulty in identification on the earlier imaging of our patient.Reference Skowronska, Kmiec, Jurkiewicz, Malczyk, Kurkowska-Jastrze and Czlonkowska 28 It is important to make this distinction with the eye-of-the-tiger sign, such as in PKAN, where there is a more central hyperintensity within the hypointense globus pallidus.Reference Hogarth, Gregory and Kruer 29 Nevertheless, this distinction is likely not absolute as several patients with MPAN have been found to have an eye-of-the-tiger sign.Reference Skowronska, Kmiec, Kurkowska-Jastrzębska and Czlonkowska 30 However, it appears that, if present, it remains less impressive in signal intensity than when found in PKAN.
In this particular case, the clinical progression together with the typical MRI findings, established the diagnosis. Although the differential diagnosis for each step of progression is broad, the diagnostic possibilities narrowed once it was identified that this was a neurodegenerative condition affecting many aspects of the nervous system. MRI pattern recognition can greatly aid in fine tuning the diagnosis in conditions where iron accumulation is present. Caution is suggested, however, given that classic MRI findings may be limited early on in the disease requiring more time for iron deposition to take place.
The NBIAs are associated with a constellation of symptoms ranging from developmental and cognitive delay to movement disorders to spastic paraplegia. MPAN, more distinctively, has been found to be strongly associated with optic atrophy, motor axonopathy as well as neuropsychiatric abnormalities. Given the broad overlap, heterogeneity of symptoms and signs and slow unraveling of the disease course, we suggest that at least the four most common NBIA disorders (PKAN, PLAN, MPAN, and BPAN) be included in testing along with the HSP panel. Hayflick et al.’s recent overview demonstrated that these four disorders make up around 85% of the NBIA spectrum suggesting a high-potential diagnostic yield.Reference Hayflick, Kurian and Hogarth 31
Ultimately, we believe that in widening the genetic panel to include these neurodegenerative disorders of the highest yield while being cognizant of the clinical and radiologic particularities highlighted in this case report, diagnostic accuracy can be improved and achieved earlier. Identifying these mutations earlier on could prove essential for both diagnostic confirmation and genetic counseling and potential future therapeutic benefit.
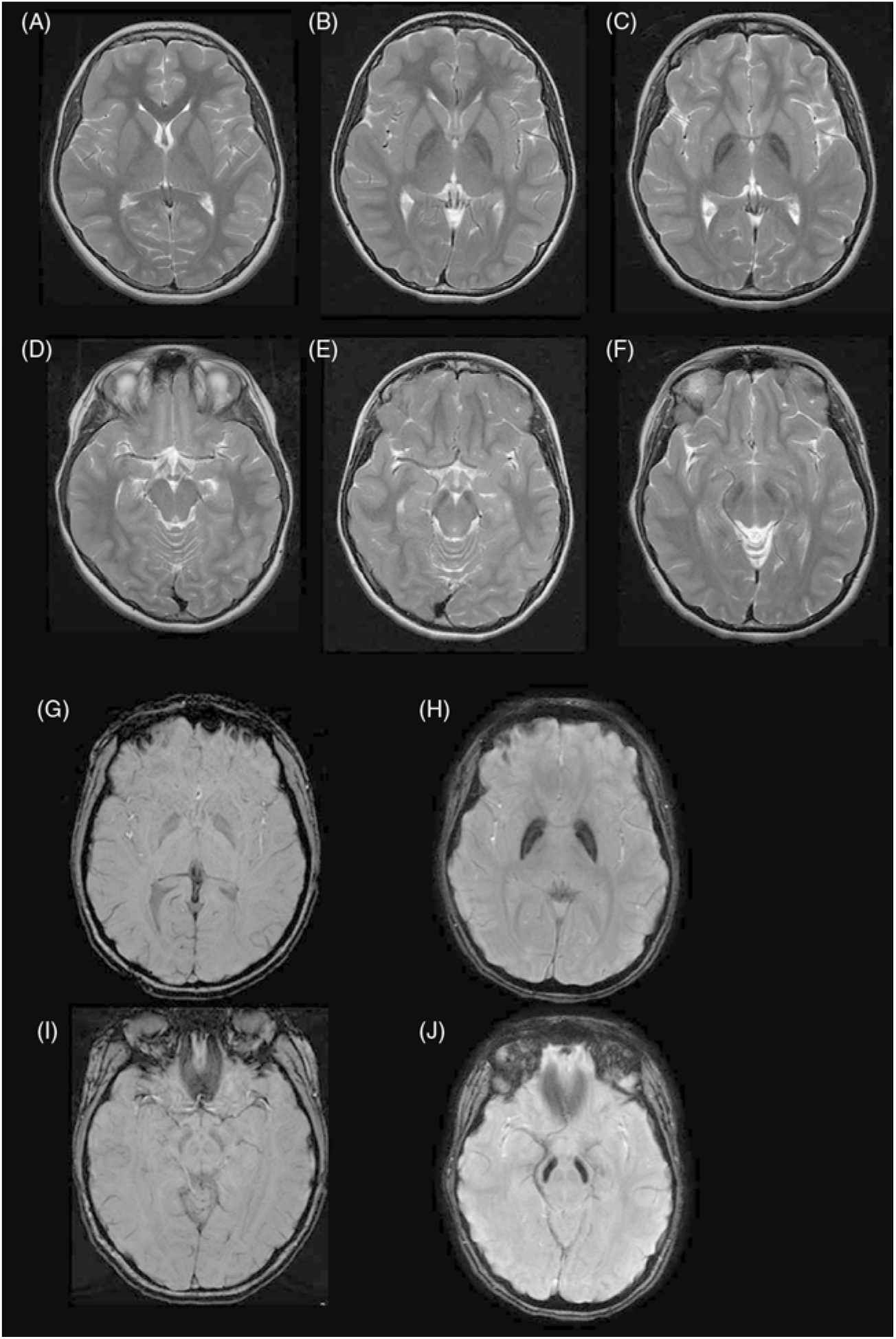
Figure 1: (A),(D): Control patient, age 13. Axial T2W images at cross section of globus pallidus (A) and substantia nigra (D). (B),(E) Patient, age 13. Axial T2W images at cross section of globus pallidus (A) and substantia nigra (E) demonstrating mild T2 hypointensity in both regions. (C),(F) Patient, age 18, with clear T2 hypointensity in the globus pallidus with a medial linear hyperintense streak (C) and T2 hypointensity in substantia nigra. (G),(I) Control age 18, Axial SWI at cross section of globus pallidus (G) and substantia nigra (I). (H),(J) Patient, age 18: Axial SWI demonstrating iron deposition in the globus pallidus with a medial linear hyperintense streak (H) and in the substantia nigra (J).
Acknowledgements
The authors would like to thank the family of the patient involved in this case report.
Conflict of Interest
There are no financial disclosures or conflicts of interests to address for all authors involved.
Statement of Authorship
SN: Wrote the manuscript, conducted the literature and chart review and performed edits.
ST: Performed chart review and performed edits. Led the article submission process.
SV: As the corresponding author, conceived the clinical case conference idea and together with the co-authors supervised and helped organize the structure of the article.

