


Hereditary hemochromatosis (HH) is a genetic disorder of iron metabolism in which progressive iron accumulation leads to pathological iron deposition and functional impairment of various organs that may become irreversible if not treated early.Reference Franchini and Veneri 1 The typical hemochromatosis patient is homozygous for the C282Y mutation in the HFE gene on the short arm of chromosome 6.Reference Franchini and Veneri 1 , Reference Datz, Haas, Rinner, Sandhofer, Patsch and Paulweber 2 In HH, iron deposition predominantly occurs in the liver, pancreas, heart, joints, gonads, and skin.Reference Franchini and Veneri 1 HH usually does not involve the nervous system, with even fewer reported cases presenting with movement disorders.Reference Jones and Hedley-Whyte 3 - Reference Williams, Vinjam, Ismail and Hassan 13 We describe three HH patients affected by isolated movement disorders without any other major systemic features of HH and review the literature with an aim to explore a possible causal relationship between HH and neurological manifestations.
Materials and Methods
The hemochromatosis clinic at London Health Sciences Centre, London, ON, Canada, was started in 1988, and a multidimensional database has been created since that time on all C282Y homozygotes. All patients were assessed by the same physician (PCA) who is a recognized expert in this field. Clinical data were collected from this database reviewed by PCA, and physical examination including neurological examination was performed. The patients in the group that had isolated neurological symptoms and possible signs involving the central nervous system (n=3) were separated from the entire group and referred to the movement disorder clinic. The next-generation sequencing (NGS) of these patients was extracted from the data performed at the London Health Sciences Clinical Molecular Genetics Laboratory using the hyperferritinemia gene panel. Testing involved NGS and copy number analysis for 15 iron-related genes (HFE2, STEAP3, SLC40A1, SLC25A38, TF, CP, HFE, TFR2, FTH1, CDAN1, B2M, HAMP, FTL, SEC23B, ALAS2) using the custom Roche Nimblegen capture-based library enrichment (Madison, WI) and Illumina NGS platform (San Diego, CA). Sequencing included all coding exons for each gene with additional 20 nucleotides of intronic sequence around each exon. This assay is clinically validated to meet the sensitivity and specificity of Sanger sequencing and multiplex ligation dependent probe amplification combined, with mean exon sequence depth of 1000× and minimum sequence depth of 100× at a single nucleotide resolution, along with custom copy number assessment of the NGS sequence data. Variants were interpreted by a certified clinical molecular geneticist (BS), and classified using the American College of Medical Genetic guidelines (PMID: 25741868). Variants classified as pathogenic, likely pathogenic, or variants of unknown clinical significance were confirmed using Sanger sequencing or multiplex ligation dependent probe amplification.
Results
Clinical details were available on 616 patients (362 males and 254 females) who were C282Y homozygotes. All patients underwent detailed hematological and biochemical workup for HH along with the genetic testing. The three patients were referred to the movement disorder clinic where they were assessed by at least one of the neurologists (NK, PR, and MJ). Examination included detailed neurological assessment including movement disorder assessment (n=3) as well as other investigations as deemed necessary to rule out other possible etiologies, including neuroimaging (brain magnetic resonance imaging [MRI] scan, n=3), electroencephalogram (n=1), copper studies for Wilson’s disease (n=3), Huntington disease (HD) gene test (n=1), liver Fibroscan (n=3), liver MRI scan (n=1), and liver biopsy (n=1). Case details for each patient follow.
Case Reports
Patient 1
This 61-year-old man of Irish and Portuguese ancestry was first diagnosed with HH at the age of 55 years. There was incidental detection of liver iron on his MRI scan when he was involved as a control subject in a research study. He had no children. There was no family history of HH. His initial serum ferritin level was 1183 µg/l with increased transferrin saturation (87.5%). Genetic testing confirmed a C282Y homozygous mutation in the HFE gene, and a heterozygous variant of unknown clinical significance c.3188C>T (p.Thr1063Met) in the codanin gene (congenital dyserythropoietic anemia-1, CDAN1) on NGS. Phlebotomy normalized his serum ferritin. Two years after being diagnosed with HH, he developed rest and action tremor and loss of dexterity in his left upper limb. His cognition was preserved. He had full extraocular movements with saccadic pursuits and slow vertical saccades. He had facial hypomimia, mild hypophonia, rigidity, and bradykinesia of the left upper and lower extremities, as well as a slow shuffling gait with reduced left arm swing. Laboratory workup showed normal complete blood count, thyroid function test, creatinine, glucose, ceruloplasmin, and 24-hour urinary copper. He had no evidence of liver involvement or diabetes mellitus. His liver Fibroscan was normal. He continued on regular phlebotomy every 3 months. His brain MRI scan showed symmetrical abnormal T1 hyperintensity in the lentiform nuclei and substantia nigra bilaterally (Figure 1). He was started on levodopa-carbidopa (100/25 mg) 1 tablet by mouth four times per day. On his follow-up 6 months later, there was improvement in his parkinsonism.
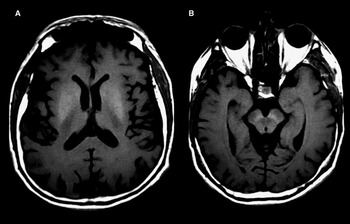
Figure 1 Brain MRI scan shows symmetrical T1 hyperintensity in the lentiform nuclei (A) and substantia nigra (B).
Patient 2
This 64-year-old man of Scottish heritage was diagnosed with HH at the age of 60 when he was detected to have increased serum ferritin (3403 µg/l) and transferrin saturation (95%) while being investigated for vertigo. Genetic testing confirmed a C282Y homozygous mutation in the HFE gene, and heterozygous pathogenic mutation c.212G>A (p.Gly71Asp) in the hepcidin gene (hepcidin antimicrobial peptide, HAMP) on NGS. His family history is significant, with his brother having HH without neurological manifestations. He was treated with phlebotomy, which normalized his serum ferritin. An otolaryngologist ruled out peripheral causes of vertigo. Echocardiography was normal, but he had a positive tilt-table test. One year after being diagnosed with HH, he developed intermittent left-sided, low-amplitude, choreiform movements primarily involving the fingers, and, occasionally, the hand and left foot. He had hypometric saccades, but no bradykinesia, rigidity, tremor, ataxia, or weakness. Laboratory workup showed normal serum electrolytes (sodium, potassium, and calcium), blood glucose, thyroid function test, creatine kinase, liver enzymes, bilirubin, creatinine, ceruloplasmin, and 24-hour urinary copper. He had decreased serum vitamin B12 that was supplemented. Genetic testing for HD was negative. Electroencephalogram was normal. Brain MRI scan showed T2 hypointensity with low signal on susceptibility-weighted imaging (SWI) in both caudate and lentiform nuclei, consistent with iron deposition (Figure 2), and to a lesser extent in bilateral dorsolateral nuclei of the thalami and dentate nuclei of the cerebellum. Incidental small asymptomatic bilateral cerebellar and left corona radiata infarcts were also seen. There was no evidence of liver involvement or diabetes mellitus. His liver Fibroscan was normal. His chorea improved on tetrabenazine 25 mg oral twice daily. He developed mild parkinsonism as a side effect of tetrabenazine. His brain MRI scan was repeated after phlebotomy treatment and was unchanged.

Figure 2 Brain MRI scan shows T2 hypointensity in both caudate and lentiform nuclei (A), and to a lesser extent in bilateral dentate nuclei of the cerebellum (C), with corresponding low signal on SWI (B and D, respectively).
Patient 3
This 66-year-old female patient was first diagnosed with HH at the age of 52 years. Her initial serum ferritin level was 1391 µg/l and transferrin saturation was 94%. She had no family history of HH. Genetic testing confirmed a C282Y homozygous mutation in the HFE gene, without non-HFE mutations on NGS. She was initially treated with phlebotomy, which was discontinued after her serum ferritin normalized. At the age of 56, she developed tremor in her head and bilateral upper extremities. There was no history of slowness, stiffness, gait, or balance abnormality. She had normal cognition and speech. On examination, she had postural and action tremor in both upper extremities with no rest tremor. There was no appendicular ataxia and her gait was normal. Her brain MRI scan showed low signal on SWI in the dentate, red nuclei and substantia nigra, consistent with iron deposition (Figure 3). Laboratory workup showed normal complete blood count, glucose, liver and thyroid function tests, creatinine, and serum ceruloplasmin. Her liver Fibroscan was slightly abnormal at 11.8 (normal <7). Liver biopsy showed a moderate parenchymal iron overload (hepatic iron concentration: 145.4 µmol/g dry weight; reference range: 0-35) with no evidence of fibrosis, consistent with HH. She resumed treatment at the age of 62 when her serum ferritin increased to 3513 µg/l and transferrin saturation reached 95.5%, with a maintenance regimen of phlebotomy every 4 months after normalization of her iron stores. Her tremor improved on propranolol 40 mg oral twice daily.

Figure 3 Brain MRI scan shows symmetrical low signal on SWI in the dentate nuclei (A), red nuclei, and substantia nigra (B).
Discussion
Iron is essential for the development and functioning of the brain, and required in myelin synthesis, neurotransmitter and enzyme function, and energy production.Reference Dusek, Jankovic and Le 14 The brain is second only to the liver among the organs with maximal physiological storage of iron.Reference Costello, Walsh, Harrington and Walsh 9 In the brain, iron distribution is not homogenous, and the globus pallidus contains the highest concentration, followed by the red nucleus, substantia nigra, putamen, and the dentate nucleus.Reference Dusek, Jankovic and Le 14 , Reference Nandar and Connor 15 Several neurodegenerative disorders, including Parkinson disease (PD), pantothenate kinase associated neurodegeneration, aceruloplasminemia, neuroferritinopathy, and Friedreich’s ataxia, are associated with increased brain iron.Reference Russo, Edwards, Andrews, O’Brien and Bhatia 7 , Reference Dusek, Jankovic and Le 14 Increased free iron, in disorders such as HH, generates free radicals that lead to cellular damage by overwhelming the detoxification system of the cell.Reference Rosana and La Rosa 10
The blood-brain barrier is thought to protect against brain iron overload.Reference Dusek, Jankovic and Le 14 The majority of HFE protein resides within the brain capillaries, choroid plexus, and ependymal cells along with transferrin receptors, and influence iron uptake by the brain.Reference Nandar and Connor 15 , Reference Connor and Lee 16 This view was supported by the postmortem finding in HH patients with iron deposition in the choroid plexus without any abnormal basal ganglia pigmentation,Reference Jones and Hedley-Whyte 3 , Reference Scully, Galbadini and McNelly 17 however, Williams et al reported postmortem iron deposition in the basal ganglia, locus coeruleus, and dentate nuclei in a case of HH with parkinsonism related to multiple system atrophy, parkinsonian type.Reference Williams, Vinjam, Ismail and Hassan 13 Iron deposition in the basal ganglia and cerebellum may cause movement disorders in HH.Reference Rutgers, Pielen and Gille 11 The reason for the susceptibility of the basal ganglia to iron deposition is not well-understood, but it may be related to selective neuronal uptake and abnormal vascular or axonal transport along white matter tracts connecting these nuclei.Reference Aquino, Bizzi and Grisoli 18 T2 hypointensity and T1 hyperintensity, as well as low signal susceptibility artifact on SWI, help detect brain iron on MRI scans.Reference Schröder and Haan 4 , Reference Nielsen, Jensen and Krabbe 5 , Reference Rosana and La Rosa 10 , Reference Rutgers, Pielen and Gille 11 , Reference Berg, Hoggenmüller and Hofmann 19 Neuroimaging in HH may show basal ganglia calcification and slight sulcal prominence on brain computed tomography,Reference Jones and Hedley-Whyte 3 T1 hyperintensity and T2 hypointensity in the basal ganglia (caudate nuclei,Reference Schröder and Haan 4 , Reference Nielsen, Jensen and Krabbe 5 globus pallidus,Reference Schröder and Haan 4 , Reference Nielsen, Jensen and Krabbe 5 , Reference Rosana and La Rosa 10 , Reference Berg, Hoggenmüller and Hofmann 19 red nuclei,Reference Schröder and Haan 4 , Reference Nielsen, Jensen and Krabbe 5 substantia nigra,Reference Schröder and Haan 4 , Reference Nielsen, Jensen and Krabbe 5 putamenReference Schröder and Haan 4 , Reference Nielsen, Jensen and Krabbe 5 , Reference Rosana and La Rosa 10 , Reference Rutgers, Pielen and Gille 11 , Reference Berg, Hoggenmüller and Hofmann 19 ) and dentate nuclei,Reference Nielsen, Jensen and Krabbe 5 , Reference Rutgers, Pielen and Gille 11 T2 hyperintensity in the striatum and white matter,Reference Demarquay, Setiey, Morel, Trepo, Chazot and Broussolle 6 and cerebellar atrophy.Reference Demarquay, Setiey, Morel, Trepo, Chazot and Broussolle 6 Brain MRI of an HH patient with multiple system atrophy, cerebellar type, showed olivopontocerebellar atrophy and cruciform hyperintensity in the pons (“hot cross bun” sign), but failed to show pathological iron deposition.Reference Di Filippo, Floridi and Rossi 12 Few cases had normal MRI scans.Reference Dethy and Caroyer 8 , Reference Costello, Walsh, Harrington and Walsh 9 , Reference Williams, Vinjam, Ismail and Hassan 13 One author describes six patients with systemic HH without neurological manifestations, but with neuroimaging findings suggestive of iron deposition.Reference Berg, Hoggenmüller and Hofmann 19 This underlines the difficulty in correlating the neurological features and brain iron deposition in HH patients. Two of our patients (patients 1 and 2) showed MRI changes suggestive of iron deposition in the basal ganglia; the third (patient 3) had iron deposition in the bilateral dentate, red nuclei and substantia nigra.
Genetic abnormalities in HFE and related proteins may affect the blood-brain barrier in few patients with HH, leaving their brain devoid of this protection.Reference Williams, Vinjam, Ismail and Hassan 13 Thus, the HFE gene mutations are considered to be the prime contributor of iron overload in the brain of HH patients,Reference Nandar and Connor 15 , Reference Beutler, Felitti, Gelbart and Ho 20 thereby increasing the risk of neurodegenerative disorders.Reference Nandar and Connor 15 , Reference Connor and Lee 16 , Reference Johnstone and Milward 21 In addition to the homozygous HFE mutation (C282Y), the NGS showed an associated mutation in the CDAN1 gene in patient 1 and HAMP gene in patient 2. The mutation in the CDAN1 gene is a rare unpublished missense variant that has a relatively low population frequency (National Heart, Lung, and Blood Institute Exome Sequencing Project: EA: A=0.06%; AA: A=0.02%). The mutation in the HAMP gene has been previously reported in hemochromatosis, but without any movement disorder or evidence of iron deposition in the brain.Reference Merryweather-clarke, Cadet and Bomford 22 It is possible that these non-HFE mutations may modify the phenotypic presentation of the HFE mutation.Reference Merryweather-clarke, Cadet and Bomford 22 Thus, HFE and non-HFE mutations leading to iron accumulation in specific subcortical brain regions may explain the unusual occurrence of movement disorders in HH.
HH usually involves the liver, pancreas, joints, skin, and gonads.Reference Franchini and Veneri 1 Neurological manifestations rarely occur in HH. Neurological involvement with movement disorders in HH patients, including parkinsonism,Reference Jones and Hedley-Whyte 3 - Reference Demarquay, Setiey, Morel, Trepo, Chazot and Broussolle 6 , Reference Dethy and Caroyer 8 , Reference Costello, Walsh, Harrington and Walsh 9 Parkinson-plus syndromes,Reference Di Filippo, Floridi and Rossi 12 , Reference Williams, Vinjam, Ismail and Hassan 13 chorea,Reference Russo, Edwards, Andrews, O’Brien and Bhatia 7 myoclonus,Reference Jones and Hedley-Whyte 3 , Reference Demarquay, Setiey, Morel, Trepo, Chazot and Broussolle 6 ataxia,Reference Jones and Hedley-Whyte 3 , Reference Nielsen, Jensen and Krabbe 5 , Reference Demarquay, Setiey, Morel, Trepo, Chazot and Broussolle 6 , Reference Dethy and Caroyer 8 , Reference Rutgers, Pielen and Gille 11 dystonia,Reference Demarquay, Setiey, Morel, Trepo, Chazot and Broussolle 6 , Reference Russo, Edwards, Andrews, O’Brien and Bhatia 7 and tremor,Reference Schröder and Haan 4 – Reference Demarquay, Setiey, Morel, Trepo, Chazot and Broussolle 6 , Reference Rosana and La Rosa 10 with mean onset age of 57 years have been reported (Table 1). Of all the previously reported patients with HH and movement disorders, two had no major systemic manifestations.Reference Dethy and Caroyer 8 , Reference Williams, Vinjam, Ismail and Hassan 13 We report three HH patients, one each with parkinsonism, chorea, and tremor, without any major systemic manifestations outside the nervous system. Metabolic disturbances in systemic HH, mainly from progressive hepatic involvement, can often produce neurological features.Reference Nielsen, Jensen and Krabbe 5 , Reference Russo, Edwards, Andrews, O’Brien and Bhatia 7 All three of our patients had normal liver function tests and there were no other metabolic abnormalities apart from the increased blood iron stores. Patient 1 had evidence of iron deposition in the liver on MRI scan, and patient 3 had evidence of iron deposition on liver biopsy. However, none of the three patients had cirrhosis.
Table 1 Reported cases of movement disorders associated with HH including our cases

ALT = alanine aminotransferase; BG = basal ganglia; CN = caudate nucleus; DBS = deep brain stimulation; DM = diabetes mellitus; DN = dentate nucleus; F = female; GP = globus pallidus; HH = hereditary hemochromatosis; HD = Huntington disease; LFT = liver function test; LN = lentiform nucleus; M = male; MSA-C = multiple system atrophy, cerebellar type; MSA-P = multiple system atrophy, parkinsonian type; OPCA = olivopontocerebellar atrophy; OH: orthostatic hypotension; P: putamen; RLS: restless legs syndrome; RN: red nucleus; SN: substantia nigra; Th = thalamus; WM = white matter.
* Liver Fibroscan = transient elastography for the measurement of liver fibrosis.
The clinical-radiological correlation between the sites of brain iron deposition and neurological presentations in few reported cases suggest a causal link between HH and movement disorders.Reference Schröder and Haan 4 , Reference Nielsen, Jensen and Krabbe 5 , Reference Rutgers, Pielen and Gille 11 The stabilization or even reversibility of movement disorders on normalization of iron stores by phlebotomy in some of the reported cases further strengthens the causal link between HH and movement disorders.Reference Schröder and Haan 4 , Reference Dethy and Caroyer 8 , Reference Rutgers, Pielen and Gille 11 However, there are other reported cases with HH and movement disorders that failed to respond to phlebotomy,Reference Jones and Hedley-Whyte 3 , Reference Nielsen, Jensen and Krabbe 5 , Reference Demarquay, Setiey, Morel, Trepo, Chazot and Broussolle 6 which may be explained by the prolonged insult to the brain making the pathology irreversible.Reference Rutgers, Pielen and Gille 11 In one report, a patient showed improvement in fatigue and headache, and the possibility of improvement in parkinsonism and ataxia was not ruled out as the serum ferritin had not reached normal levels at the time of reporting the case.Reference Nielsen, Jensen and Krabbe 5 In another report, normalization of iron stores with phlebotomy led to reversal of brain MRI scan findings but the patient’s parkinsonism and tremor improved only with deep brain stimulation of the right ventral intermediate nucleus of thalamus.Reference Rosana and La Rosa 10 All three of our patients improved with symptomatic drug therapy. Their failure to respond to normalization of iron stores with phlebotomy may be explained by the probably irreversible brain pathology resulting from the prolonged insult during the years of exposure.
The relatively high frequency of HH in Caucasians (0.3%)Reference Datz, Haas, Rinner, Sandhofer, Patsch and Paulweber 2 , Reference Steinberg, Cogswell and Chang 23 highlights the possibility that patients with HH may coincidentally develop other neurodegenerative movement disorders such as idiopathic PD and essential tremor.Reference Demarquay, Setiey, Morel, Trepo, Chazot and Broussolle 6 , Reference Russo, Edwards, Andrews, O’Brien and Bhatia 7 Few authors have reported a positive correlation between HH and idiopathic PD.Reference Costello, Walsh, Harrington and Walsh 9 , Reference Dekker, Giesbergen and Njajou 24 , Reference Guerreiro, Bras and Santana 25 However, larger studies have shown no correlation between HFE mutation and the risk of idiopathic PD,Reference Buchanan, Silburn, Chalk, Le Couteur and Mellick 26 - Reference Biasiotto, Goldwurm and Finazzi 28 and one group suggested the presence of the C282Y HFE mutation may be protective against the development of idiopathic PD.Reference Buchanan, Silburn, Chalk, Le Couteur and Mellick 26 The parkinsonism in patient 1 was asymmetrical and resembled idiopathic PD. However, its development in the setting of pathological brain iron deposition indicates a secondary cause related to HH. The initial improvement of parkinsonism with levodopa can also occur in secondary parkinsonism, such as vascular parkinsonismReference Demirkiran, Bozdemir and Sarica 29 and Parkinson-plus syndromes such as multiple system atrophy.Reference Jankovic 30 Thus, a trial of levodopa for parkinsonism associated with HH is indicated. The long-term response to levodopa in this case, however, is unpredictable.
Other movement disorders have also been reported in HH. We describe, for the first time, chorea associated with HH. Chorea was reported in one patient with HH who was later confirmed to have HD, however.Reference Russo, Edwards, Andrews, O’Brien and Bhatia 7 The chorea in patient 2 was most likely attributed to iron deposition in the caudate nuclei. The postural and action tremor in patient 3 resembled essential tremor. However, pathological brain iron deposition in the bilateral dentate nuclei, red nuclei, and substantia nigra suggests her tremor being likely secondary to HH.
Conclusion
We present three unique cases of HH associated with different movement disorders. Although two of them had evidence of iron overload in their liver, none of our patients had any major systemic manifestations. The availability of NGS helps to identify non-HFE mutations in HH patients, which may act as genetic modifiers of the phenotypic presentation of HFE mutations. We report the first case of chorea associated with HH. Patient 1 developed parkinsonism as the initial presentation of HH, which was levodopa-responsive. Patient 3 presented with head and bilateral upper extremity tremor responsive to propranolol.
The clinical-radiological correlation between the sites of brain iron deposition and neurological manifestations is demonstrated in our HH patients. The literature supports iron deposition in the basal ganglia and cerebellum on brain pathology in HH. Finally, the stabilization or reversal of neurological features on normalization of iron stores by phlebotomy in few reported cases indicates that the neurological involvement in HH appears to be more than probabilistic. We propose the term “neuro-hemochromatosis” for HH patients presenting with predominant neurological features (movement disorders in our cases) in the absence of any major systemic manifestations of HH. HH should be considered in the differential diagnoses of movement disorders with pathological iron deposition in the brain, because normalization of iron stores early in the disease process may result in complete recovery or stabilization of the neurological manifestations, including movement disorders. In addition to phlebotomy, appropriate drug treatment depending on the phenotype of movement disorders should be considered.
Disclosures
NK, PR, BS, and PCA report no disclosures relevant to this manuscript. MJ receives speaker and consultant honoraria from Merz Pharmaceuticals, Allergan, AbbVie; research grants from CIHR, AMOSO, Allergan, Merz Pharmaceuticals, and Lawson Health Research Institute; is part of the AGE-WELL Network of Centers of Excellence (NCE) of Canada program; and from time to time, serves on advisory boards of Allergan, Boston Scientific, AbbVie, and Merz Pharmaceuticals.
Statement of Authorship
NK undertook the conception, design, and writing the first manuscript. PR Dr. Rizek undertook the conception, review, and critique of the article. BS undertook sequencing and interpretation. PCA undertook the review and critique. MJ undertook the review and critique.




