


Introduction
Glioblastoma (GBM) is the most malignant primary brain tumor and accounts for nearly 15% of all central nervous system (CNS) tumors, while astrocytic and oligodendroglial tumors (diffuse and anaplastic) account for approximately 7% of all primary CNS tumors.Reference Ostrom, Gittleman, Truitt, Boscia, Kruchko and Barnholtz-Sloan1 In diffuse gliomas, survival outcomes, response to treatment, and clinical characteristics of patients vary by the tumor grade and molecular alterations.Reference Stupp, Mason and van den Bent2 While the 2007 World Health Organization Classification of Tumours of the Central Nervous System largely separated glioma types based on histological featuresReference Louis, Ohgaki and Wiestler3, the newest revision (from 2016) integrated molecular markers such as isocitrate dehydrogenase (IDH) 1 and 2 mutations, TP53 mutations, ATRX loss, and 1p/19q codeletion in classifying diffuse gliomas.Reference Louis, Perry and Reifenberger4
Critical gene alterations leading to glioblastomas have been attributed to three pathways with varying degrees of frequencies. These include the RB1 pathway (68%), the TP53 (64%), and P13K/PTEN pathway (50%).Reference Parsons, Jones and Zhang5 Glioblastomas can develop de novo, without lower grade precursors (primary GBM) or they can develop through the malignant transformation of lower grade tumors (secondary GBM). Grade II or III astrocytomas can transform into secondary GBM and diffuse oligodendrogliomas can transform to anaplastic oligodendrogliomas (AOs).Reference Cohen, Holmen and Colman6 It is not possible to distinguish secondary from primary glioblastomas by histology alone.Reference Yan, Parsons and Jin7 Approximately 90% of glioblastomas are primary and 10% are secondary.Reference Appin and Brat8 There are distinct molecular identities for primary and secondary GBMs, astrocytomas, and oligodendrogliomas based on various coexisting mutations.
Over the last decade, the advent of DNA methylation studies and increasing knowledge of specific genetic alterations in glioma genomes has created the potential for classifying diffuse gliomas based on molecular features rather than histopathology alone. This paper reviews survival outcomes and clinical characteristics of patients based on IDH1/2 and other mutations typically seen in diffuse gliomas and based on DNA methylation studies. The classification system suggested by recent DNA methylation studies and glioma molecular markers will be discussed along with those of the current WHO 2016 tumor classification system.
Removal of Previous Classification of Oligoastrocytoma
Historically, a high degree of interobserver variability was noted in the diagnosis of gliomas,Reference Coons, Johnson, Scheithauer, Yates and Pearl9 likely as a byproduct of primarily histological diagnosis rather than molecular grouping. Astrocytomas are known to harbor concurrent TP53 mutations (50–90%) and ATRX mutations (loss of function mutations), while oligodendrogliomas have 1p/19q codeletion (50–70%), normal ATRX, and a much lower number of TP53 mutations (5–10%).Reference Kim and Liau10,Reference Ueki, Nishikawa and Nakazato11 Prior to the most recent WHO brain tumour classifications system, oligoastrocytomas were defined as mixed Grade II gliomas.Reference Louis, Ohgaki and Wiestler3 Maintz et al. showed that oligoastrocytomas without 1p/19q codeletion often had TP53 mutations.Reference Maintz, Fiedler and Koopmann12
The Cancer Genome Atlas Research Network used integrative genomic analysis to find three distinct genetic subtypes of low-grade gliomas which were nonoverlapping and of prognostic significance. The first subtype included tumors with IDH mutations and 1p/19q codeletions, and was found to have the best clinical outcome. The second group was IDH-mutated tumors, but with absent 1p/19q codeletions. The third group was IDH wildtypes and exhibited poor clinical outcomes, similar to that of primary glioblastoma.Reference Brat, Verhaak and Aldape13
The 2016 WHO Classification of Tumours of the Central Nervous System recommends that a diagnosis of oligoastrocytoma, not otherwise specified (NOS) and anaplastic oligoastrocytoma, NOS, be used only in exceptional situations involving gliomas exhibiting both astrocytic and oligodendroglial components that cannot be subjected to molecular testing or those with inconclusive molecular findings.Reference Reifenberger, Collins, Hartmann, Louis, Ohgaki, Wiestler and Cavenee14
IDH Mutations
Role in Gliomagenesis
Mutations in IDH enzymes have been implicated in some cancers such as acute myeloid leukemia, cholangiocarcinoma, myelodysplastic syndromes, and gliomas to name a few.Reference Jin, Hu and Yu15,Reference Dang, Yen and Attar16 Of the three isozymes (IDH1, IDH2, and IDH3) which exist, IDH1 is in the cytosol and peroxisome while IDH2 and IDH3 are in the mitochondria.Reference Dang, Yen and Attar16,Reference Choi, Ganji and DeBerardinis17 IDH mutations are typically heterozygous and involve arginine sites involved with the binding of isocitrate (namely the R132 in IDH1 and R172 and R140 codons in IDH2).Reference Dang, Yen and Attar16,Reference Gross, Cairns and Minden18 They result in the conversion of alpha ketoglutarate (KG) to D-2-hydroxyglutarate (2-HG), which accumulates to generate epigenetic dysregulation through the inhibition of alpha KG-dependent histone and DNA methylases, thereby blocking cellular differentiation and promoting tumorigenesis.Reference Parsons, Jones and Zhang5,Reference Dang, Yen and Attar16,Reference Gross, Cairns and Minden18,Reference Yamashita, da Costa Rosa, Borodovsky, Festuccia, Chan and Riggins19
IDH mutations can occur in any grade of glioma (most frequently in Grade II/III astrocytomas and oligodendrogliomas as well as secondary glioblastomas) but are not seen in pilocytic astrocytomas.Reference Louis, Perry and Reifenberger4 Most tumors with 1p/19q codeletions have also been shown to have IDH mutations.Reference Metellus, Coulibaly and Colin20 IDH1 mutations occur very early in gliomagenesis, even prior to TP53 mutations and 1p/19q codeletions.Reference Watanabe, Nobusawa, Kleihues and Ohgaki21–Reference Wang, Tang and Liang23 Tumors without IDH1 mutation might instead have IDH2 R172 mutation.Reference Yan, Bigner, Velculescu and Parsons22 IDH1 and IDH2 mutations are in 80–88% of oligodendrogliomas.Reference Hartmann, Meyer and Balss24 More than 70% of astrocytomas and oligodendrogliomas (WHO Grade II and Grade III) harbor the IDH1 R132 mutation.Reference Yan, Parsons and Jin7 If the R132H mutation-specific antibody (most common) testing is negative, the patient may have a noncanonical mutation, which can be determined with DNA sequencing.Reference Aldape, Nejad, Louis and Zadeh25 IDH mutations are linked with the proneural phenotype and secondary glioblastomas, as well as increased DNA methylation (called G-CIMP for glioma CpG island methylation phenotype).Reference Cohen, Holmen and Colman6
In contrast to adult gliomas, pediatric gliomas do not typically harbor IDH mutations.Reference Rizzo, Ruggiero, Martini, Rizzo, Maurizi and Riccardi26 In a study of 43 patients with high-grade gliomas, Pollack et al. found that 35% of children over the age of 14 years had an IDH1 mutation but those who were younger than this group had no IDH mutations.Reference Pollack, Hamilton and Sobol27 This suggests that pediatric gliomas may be unique molecular entities that differ markedly from adult gliomas.
IDH1 Versus IDH2 Mutations
In gliomas, IDH1 mutations are generally more common than IDH2 mutations and are known to be mutually exclusive.Reference Wang, Tang and Liang23 In their genomic analysis of GBM patients, Parsons et al. found that 12% of primary GBM patients had an IDH1 mutation and Wang et al. found that 14.1% of primary GBM patients had either IDH1/2 mutations.Reference Parsons, Jones and Zhang5,Reference Wang, Tang and Liang23 Hartmann et al. studied 1010 cases of diffuse gliomas and found that 73% of diffuse astrocytomas had an IDH1 mutations (compared to 0.9% with IDH2 mutations) and 64% of anaplastic astrocytomas (AAs) had IDH1 mutations (compared to 0.8% with IDH2 mutations). In the AO group, 69.5% had IDH1 mutations while 5.2% had IDH2 mutations. There were IDH1 mutations in 82% and IDH2 mutations in 4.7% of diffuse oligodendrogliomas. Of note, this study was performed prior to the newest WHO brain tumor classification (2016) and thus a separate group of “oligoastrocytomas” was also discussed in the original article. The IDH2 mutations were highly associated with 1p/19q codeletions, as per a recent study by Wang et al., who found that 44.4% of IDH2-mutated gliomas had a 1p/19q codeletion. In addition, IDH2 mutations in gliomas are mutually exclusive to other mutations typically seen in gliomas (PTEN, TP53, ATRX).Reference Wang, Tang and Liang23
Clinical Significance of IDH Mutations in Gliomas
In a study by Yan et al., patients with IDH-wildtype GBMs were reported to be older than their IDH-mutated counterparts (59 versus 32 years) and are thought to have worse prognosis; similarly, IDH-wildtype AA patients were older than IDH-mutated AA patient counterparts (56 versus 34 years).Reference Yan, Parsons and Jin7 With regard to treatment response, Li et al. suggested that gliomas with IDH mutations are more sensitive to radiation therapy while other studies suggested that IDH-mutated gliomas may have a higher response to alkylating agents.Reference Li, Chou and Chen28–Reference Houillier, Wang and Kaloshi30
Grade II–IV gliomas with IDH mutations have better survival than those without, suggesting the prognostic significance of this mutation, independent of other factors such as tumor grade and O6-methylguanine-DNA-methyltransferase (MGMT) methylation.Reference Sanson, Marie and Paris31 This trend for improved survival with IDH-mutated gliomas when compared to their non-IDH-mutated counterparts was also noted by several other studies in gliomas.Reference Yan, Parsons and Jin7,Reference Hartmann, Meyer and Balss24,Reference Wick, Hartmann and Engel32–Reference Weller, Felsberg, Hartmann and Berger34 Weller et al. demonstrated a progression-free survival benefit in GBMs with IDH mutations compared to those with IDH-wildtype status (16.2 versus 6.5 months) and a trend toward benefit for overall survival (30.2 versus 11.2 months).Reference Weller, Felsberg, Hartmann and Berger34 Though there may be a progression-free survival benefit for IDH2 mutations when compared to IDH1 mutations, overall survival is not thought to be different between the two groups of mutations.Reference Wang, Tang and Liang23 Several other studies have shown that IDH1 and IDH2 mutations predict longer survival in secondary glioblastomas and Grade II and Grade III gliomas.Reference SongTao, Lei and Si29,Reference Houillier, Wang and Kaloshi30 Parsons et al. found that GBM patients with IDH1 mutations had an improved overall survival of 3.8 years (45.6 months) compared to the 1.1 years (13.2 months) noted in those without IDH1 mutations. The survival benefit was noted independent of an additional TP53 mutation.Reference Parsons, Jones and Zhang5 IDH mutations were associated with improved median survival for both GBM (median overall survival [OS] of 31 versus 15 months in those who were IDH wildtype) and AA (median OS of 65 versus 20 months without mutations).Reference Yan, Parsons and Jin7
In summary, IDH mutations occur early in gliomagenesis and most commonly occur in low-grade and AAs and oligodendrogliomas as well as in secondary glioblastomas. Though multiple studies have addressed the possibility of classifying gliomas by their IDH status (rather than by histology and IDH status, as per current WHO guidelines), there is no clear evidence that IDH status alone determines tumor grade as prognosis is also dependent on other concurrent mutations associated with gliomas. These mutations will be discussed in the next section.
Other Common Genetic Alterations in Diffuse Gliomas
IDH-wildtype GBMs have higher rates of PTEN, EGFR, CDKN2A, or CDKN2B mutations while IDH-wildtype low-grade gliomas can have EGFR amplification, TERT (telomerase reverse transcriptase) mutations, and BRAF mutations (Table 1).Reference Aibaidula, Chan and Shi35 Of note, while H3F3A mutations have been shown to confer worse prognosis, Aibaidula et al.Reference Aibaidula, Chan and Shi35 found that they occur in 9.5% of low-grade gliomas but are not relatively as common in IDH-wildtype glioblastomas. The most common genetic abnormalities in IDH-wildtype glioblastomas are TERT mutations (72–90%), EFGR amplifications (35–45%), homozygous deletion of CDKN2A/CDKN2B (60%), loss of chromosomes 10p or of 10q (50% and 70%, respectively), TP53 mutation (28–35%), and PTEN mutations (25–35%).Reference Louis, Perry and Reifenberger4 In contrast, IDH-mutant GBMs are more often associated with a hypermethylator phenotype, originate from a proneural transcriptional profile, and have typical genetic alterations such as TP53 (81%), ATRX mutation (71%), loss of chromosome arm 19q (50%), loss of chromosome arm 10q (>60%), and TERT mutations (30%).Reference Louis, Perry and Reifenberger4 In addition, IDH-mutant GBMs often lack EGFR amplification, which is more consistent with secondary glioblastomas.Reference Weller, Felsberg, Hartmann and Berger34,Reference Verhaak, Hoadley and Purdom36 EGFR amplification and PTEN mutations are extremely rare in the IDH-mutated GBM population and TERT mutations are less frequently seen (26%).Reference Louis, Perry and Reifenberger4 In lower grade gliomas (IDH wildtype), Aibaidula et al. found that 26.8% of cases had TERT, 13.8% had EFGR amplification, 6.9% had BRAF mutations, and 9.5% had H3F3A mutations.Reference Aibaidula, Chan and Shi35 EGFR alterations and PTEN deletions are more commonly seen in primary GBMs than secondary GBMs.Reference Appin and Brat8
Table 1: Common mutations seen in IDH-wildtype low-grade gliomas versus IDH-wildtype glioblastomas

However, there are rare histological variants of IDH-wildtype GBMs whose genetic profiles differ from the typical IDH-wildtype GBMs. Giant cell glioblastomas are characterized by bizarre-looking multinucleated giant cells. While this variant frequently possesses PTEN and TP53 mutations, they typically lack EGFR amplification/overexpression and homozygous CDKN2A deletion.Reference Ohgaki, Kleihues, Plate, Nakazato, Bigner, Louis, Ohgaki, Wiestler and Cavenee37 Their clinical prognosis is fairly better compared to the more typical IDH-wildtype GBM.Reference Ohgaki, Kleihues, Plate, Nakazato, Bigner, Louis, Ohgaki, Wiestler and Cavenee37 Another variant of IDH-wildtype GBM is made up of closely packed epithelioid cells, aptly named epithelioid glioblastoma. These are aggressive tumors, even compared to an ordinary GBM and about 50% of its population contain BRAF V600E mutation.Reference Ellison, Kleinschmidt-DeMasters, Park, Louis, Ohgaki, Wiestler and Cavenee38 The third IDH-wildtype variants are gliosarcomas, which are characterized by alternating areas of glial and mesenchymal differentiation. They contain the typical GBM PTEN mutations, CDKN2A deletions, and TP53 mutations, but only have infrequent EGFR amplifications. Gliosarcomas have the same outcome as a classic GBM, but systemic metastasis and skull invasions had been reported for this variant.Reference Burger, Giangaspero, Ohgaki, Biernat, Louis, Ohgaki, Wiestler and Cavenee39
1p/19q Codeletion
In the 2016 WHO classification update, oligodendrogliomas are molecularly defined by IDH mutations with concurrent codeletion of chromosomes 1p and 19q. They often have TERT mutations but ATRX loss is not present. Testing of 1p/19q codeletion is often completed through FISH (fluorescence in situ hybridization), among other methods of testing currently available.Reference Masui, Mischel and Reifenberger40 However, detection of 1p/19q codeletion by FISH is not always accurate and can lead to false positives (e.g., when FISH is positive in the event of interstitial deletions involving the areas with probe hybridization or with monosomy, without complete deletion of the 1p and 19q chromosomes).Reference Ballester, Huse, Tang and Fuller41 This potential for false classification using FISH testing for this codeletion could be corrected using more detailed testing such as chromosomal microarray.Reference Ballester, Huse, Tang and Fuller41 In addition, 1p/19q codeletion can also be found in glioblastomas and astrocytic tumors, presenting a diagnostic challenge. DNA methylation studies could assist in properly classifying tumors in these cases (discussed in a separate section). The presence of this chromosomal codeletion of 1p and 19q has been shown to confer a significant survival advantage in patients who were treated with radiotherapy and Procarbazine, Lomustine, Vincristine (PCV) chemotherapy.Reference Taal, van der Rijt and Dinjens42,Reference van den Bent43
ATRX Mutations
If a tumor is negative for a 1p/19q codeletion in the setting of IDH-wildtype status, it is likely an IDH-wildtype AA or glioblastoma.Reference Vogazianou, Chan and Bäcklund44 In contrast, astrocytomas also have IDH mutations but with loss of ATRX – they do not typically have TERT promotor mutations or 1p/19q codeletions.Reference Louis, Perry and Reifenberger4 ATRX mutations lead to lack of expression of ATRX and are typically associated with IDH1/2 mutations and TP53 mutations in Grade II and III astrocytomas and IDH-mutated GBM.Reference Louis, Perry and Reifenberger4 ATRX retention with 1p/19q codeletion confers a better prognosis in anaplastic gliomas.Reference Louis, Perry and Reifenberger4 Wiestler et al. reported better progression-free survival in IDH-mutated astrocytic tumors with ATRX loss (55.6 months) compared to those without ATRX loss (31.8 months).Reference Wiestler, Capper and Holland-Letz45
TP53 Mutations
TP53 is a tumor suppressor gene and mutations lead to overexpression of p53 as a gain-of-function mechanism.Reference Louis, Perry and Reifenberger4 A high frequency of TP53 mutations in GBM patients with IDH1 mutations has been reported.Reference Parsons, Jones and Zhang5 Cohen et al. found that 80% of IDH-mutated AAs and IDH-mutated glioblastomas have TP53 mutations.Reference Cohen, Holmen and Colman6 These mutations are also present in diffuse astrocytic tumors but are not prognostic of clinical outcomes.Reference Gillet, Alentorn and Doukouré46 In contrast, only 9% of AOs have been reported to have TP53 mutations.Reference Yan, Parsons and Jin7 An update in 2018 by the Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy (cIMPACT-NOW) recommended that an IDH-mutated Grade II or III astrocytoma can be diagnosed in the setting of an astrocytic-looking tumor that expressed IDH mutation, loss of ATRX, and/or a diffuse p53 positivity, even when 1p/19q testing is absent.Reference Louis, Giannini and Capper47 This update allows for further improvements in the classification of astrocytomas since the 2016 WHO criteria.
TERT (Telomerase Reverse Transcriptase)
TERT promoter mutations increase promoter activity and are seen in glioblastomas (up to 80%) and oligodendrogliomas. These mutations are rarely found in astrocytomas or secondary (IDH-mutated) glioblastomas but are often found in IDH-wildtype glioblastomas.Reference Louis, Perry and Reifenberger4 TERT promoter mutations are inversely correlated with frequency of IDH and TP53 mutations in glioblastomas.Reference Nonoguchi, Ohta, Oh, Kim, Kleihues and Ohgaki48 TERT promotor mutations and ATRX mutations were found to be mutually exclusive in most cases.Reference Eckel-Passow, Lachance and Molinaro49 TERT mutations are often associated with aggressive clinical course in a diffuse astrocytic gliomas. The 2018 c-IMPACT-NOW update suggests that it is best to test for IDH mutation and 1p/19q codeletion status to rule out the diagnosis of oligodendroglioma as most oligodendrogliomas are known to exhibit TERT promoter mutation as well.Reference Brat, Verhaak and Aldape13 Age at time of diagnosis, molecular markers, and grade of tumor are independent associations with overall survival.Reference Eckel-Passow, Lachance and Molinaro49 Patients with GBM who have TERT and IDH mutations do not have better survival than those with TERT mutations only, suggesting that IDH mutations do not dictate prognosis alone and that TERT mutations hold independent prognostic value.Reference Eckel-Passow, Lachance and Molinaro49,Reference Aibaidula, Chan and Shi35,Reference Simon, Hosen and Gousias50
Exploring the Role of Tumor Stratification Based on IDH Mutation Status
Several studies have suggested that histologically Grade II and Grade III tumors may have a dismal prognosis if associated with IDH-wildtype status.Reference Yan, Parsons and Jin7,Reference Metellus, Coulibaly and Colin20,Reference Eckel-Passow, Lachance and Molinaro49 Metellus et al. studied 47 low-grade gliomas (18 oligodendrogliomas, 7 astrocytomas, and 22 oligoastrocytomas – as per the WHO 2007 classification system) and found that absence of IDH1/2 mutations, older age, lower extent of surgery, and frontotemporoinsular tumors with a highly infiltrative pattern were poor prognostic factors. These authors found that low-grade gliomas with IDH1/2 mutations had a median PFS of 4.7 years (56.4 months), while IDH1/2-wildtype tumors had median progression free survival (PFS) of 1.4 years (16.8 months). Reuss et al. Reference Reuss, Kratz and Sahm51 analyzed 160 IDH-wildtype Grade II (40/160) and Grade III (120/160) astrocytomas (by WHO 2007 classification system) and found that 78% of these tumors could be reclassified as glioblastomas by molecular analysis, while 17% had a molecular profile consistent with GBM-H3F3A or GBM-H3-K27 subtypes. Molecular subtypes in this study were determined by G-CIMP phenotype testing and determination of copy number alterations on 450k methylation analysis as well as sequencing for IDH1/2, H3F3A, and TERT promoter mutations. Interestingly, of the 78% of tumors reclassified as GBMs (as per hallmark genetic alterations, DNA methylation profiles, and copy number profiles), most also had at least a TERT promoter mutation, 10q loss and 7q gain, and EGFR amplification (findings often associated with IDH-wildtype GBMs). True to the molecular diagnosis, the median survival for the molecular GBM subgroup was approximately 19.4 months. In summary, the authors found that most IDH-wildtype astrocytomas determined by histology could be reclassified as GBMs by molecular basis and have a similar median overall survival to conventional GBMs.
Other studies contradict this and suggest that the IDH status alone does not supersede the histological grading criteria as the presence of several associated mutations also contributes to the overall survival.Reference Aibaidula, Chan and Shi35,Reference Chan, Yao and Zhang52 Chan et al. Reference Chan, Yao and Zhang53 studied 214 lower grade gliomas and found that median overall survival was most favorable for those with IDH1/2 mutation along with either 1p/19q codeletion or a TERT mutations, while it was least favorable for those with IDH-wildtype status and a TERT mutation or EGFR amplification. However, they found that the histological grade in these cases was still more representative of survival outcomes than the mutations in tumors within each grade. Aibaidula et al. analyzed 718 Grade II and Grade III gliomas (per WHO 2007 classification) and found that a molecularly higher grade of tumors (IDH-wildtype, EGFR, H3F3A, or TERT mutations) had median overall survival of 1.23 years (14.76 months), whereas the “molecularly lower grade” tumors (IDH mutated with no EGFR, TERT, or H3F3A mutations) had a significantly higher overall survival of 7.63 years (91.56 months). These tumors had higher survival than the approximately 30-month overall survival reported in IDH-mutant glioblastomas who undergo standard of care chemotherapy.Reference Weller, Felsberg, Hartmann and Berger34 A more recent meta-analysis by Vuong et al. also demonstrated a propensity for IDH-wildtype low-grade gliomas to have a poor prognosis when associated with TERT promoter mutations, EGFR amplifications, and H3F3A mutations, whereas ATRX mutation, 7q gain/10q loss, and CDKN loss did not influence prognosis.Reference Vuong, Tran and Ngo54
Thus, there is not enough evidence that IDH status alone should dictate the classification of glioma; instead, IDH status must be taken in conjunction with other prognostic mutations commonly found in gliomas in addition to the clinical and radiological status of the patient. This is reflected in the cIMPACT-NOW consortium guidelines (2018) which designate IDH-wildtype Grade II or III tumors, with one of the following molecular characteristics: EGFR amplification, or combined whole chromosome 7 gain and whole chromosome 10 loss, or TERT promoter mutation, as “diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV.”Reference Brat, Aldape and Colman55 The clinical implications of this new designation mean that a combination of chemotherapy and radiotherapy is recommended for these types of tumors. In addition, inclusion criteria for clinical trials may be extended to include this group of patients.Reference Brat, Aldape and Colman55
BRAF fusions versus mutations
BRAF is an intracellular serine/threonine kinase component of the mitogen-activated protein kinase pathway. It is involved in the activation of transcription factors that induces cell proliferation, survival, and tumorigenesis – it can also trigger differentiation and senescence.Reference Horbinski56,Reference Cantwell-Dorris, O’Leary and Sheils57 BRAF is the more potent activator of MEK compared to other RAF isoforms and has a greater oncogenic potential. The most frequent BRAF mutations found in gliomas are BRAF gene rearrangements and fusions.Reference Horbinski56
BRAF fusions
A tandem duplication and rearrangement on chromosome 7q34 between BRAF and a gene, KIAA1549, produces a fusion gene.Reference Horbinski56 KIAA1549:BRAF fusion is detected in about 80% of pediatric pilocytic astrocytomas, but rarely in low-grade diffuse astrocytomas.Reference Becker, Scapulatempo-Neto and Carloni58,Reference Jeuken and Wesseling59 Because pilocytic astrocytomas frequently harbor BRAF fusions, it has been suggested that along with other markers, the presence of this fusion may help support the diagnosis of pilocytic astrocytoma.Reference Collins, Jones and Giannini60 The presence of the BRAF fusion gene was shown to be associated with a better overall survival in pilocytic astrocytomas.Reference Becker, Scapulatempo-Neto and Carloni58 Despite its uncertain diagnostic value, the detection of KIAA1549:BRAF fusion may be useful in identifying low-grade astrocytic glioma patients with a longer survival outcome.Reference Jeuken and Wesseling59
BRAF V600E mutation
BRAF V600E mutations account for 90% of mutations in the BRAF gene.Reference Cantwell-Dorris, O’Leary and Sheils57 The V600E mutation was found in two-thirds of pleomorphic xanthoastrocytomas (PXA), and in approximately 20% of gangliogliomas in a series of 1320 adult and pediatric tumor.Reference Schindler, Capper and Meyer61 Similar results were found by Horbinski, with BRAF V600E mutations being found in 73.8% of PXAs and 19.2% of gangliogliomas.Reference Horbinski56 A high percentage of epithelioid GBMs (7/13, 53.8%) also harbor V600E mutation. Six out of the seven BRAF-mutated epithelioid GBM cases were observed in adults.Reference Kleinschmidt-DeMasters, Aisner, Birks and Foreman62 Despite its high frequency in epithelioid GBMs, only 7.7% of adult GBMs showed BRAF V600E immunopositivity.Reference Dahiya, Emnett and Haydon63 Despite the high incidence of BRAF V600E mutations in the tumor groups mentioned earlier, it is important to keep in mind that the absence of this mutation does not exclude a histological diagnosis of these tumors.Reference Kleinschmidt-DeMasters, Aisner, Birks and Foreman62
Histone mutations
The 2016 WHO classification identified a new entity called diffuse midline glioma, H3 K27M mutant.Reference Louis, Perry and Reifenberger4 Histone H3 subgroups include G34R/V mutations (seen in pediatric high-grade gliomas of the cerebral hemispheres) and the K27M mutations (seen in the midline pediatric high-grade gliomas).Reference Mackay, Burford and Molinari64 G34R/V histone mutations tend to occur in the cerebral hemispheres (not midline) and are mutually exclusive to the more common H3.3 K27M mutation.
H3 K27M mutations can be found in pediatric and adult diffuse intrinsic pontine gliomas (DIPG), adult diffuse midline gliomas (located in the thalamus, spinal cord, pons, and brainstem), gangliogliomas, and pilocytic astrocytomas.Reference Kleinschmidt-DeMasters and Mulcahy Levy65,Reference Solomon, Wood and Tihan66 DIPGs were shown to have a median overall survival of 11.1 months and thalamic diffuse midline gliomas have been shown to have 10.8 month overall median survival.Reference Castel, Philippe and Kergrohen67
Several variations in histone mutations have been reported. Castel et al. have recently also found two subgroups of H3 K27M-mutant tumors, H3.1 K27M (HIST1H3B gene) and H3.3 K27M (H3F3A gene), which had distinct DNA methylation profiles, though prognostic difference between these two subtypes was not discussed in this paper.Reference Castel, Philippe and Kergrohen67 K27M mutations in H3F3A or HIST1H3B genes have been shown in other studies to confer a worse prognosis for patients with midline gliomaReference Karremann, Gielen and Hoffmann68,Reference López, Oberheim Bush, Berger, Perry and Solomon69 , irrespective of age, histological grading, or tumor morphology.Reference Kleinschmidt-DeMasters and Mulcahy Levy65 The H3F3A-mutated genotype demonstrated lower responses to radiotherapy increased metastatic spread and had earlier relapses when compared to the HIST1H3B-mutated genotype.Reference Castel, Philippe and Calmon70 The presence of an H3 G34 mutation in a diffuse astrocytic glioma indicates a high-grade biology, regardless of histological grade.Reference Ballester, Huse, Tang and Fuller41 As further research is conducted on the significance of histone mutations, there may be a role for further classifying midline gliomas based on these molecular aberrations.
In summary, distinct concurrent mutations and molecular profiles are often associated with IDH-wildtype and mutant gliomas with varying grades as described above. Presence of 1p/19q codeletion confers a significant survival advantage in patients treated with radiation and chemotherapy. Similarly, a survival advantage has been shown in astrocytic tumors with IDH mutations and ATRX loss compared to those without ATRX loss. Mutations in the tumor suppressor gene, TP53, are common in IDH-mutated astrocytomas and IDH-mutated glioblastomas but are not of any known prognostic significance. TERT promoter mutations are inversely correlated with the presence of IDH and TP53 mutations and are thus more commonly seen in IDH-wildtype glioblastomas. TERT promoter mutations are also mutually exclusive to ATRX mutations and are thus not often seen in astrocytomas. BRAF mutations and fusions can be found in many tumors. In pilocytic astrocytomas, BRAF fusions are seen in 80% of tumors and confer a survival advantage. The BRAFV600E mutation accounts for 90% of BRAF gene mutations and is found in two-thirds of PXA and about half of epitheliod glioblastomas. Several variations of histone mutations have been reported thus far. H3 K27M mutation, which confers a survival disadvantage, has been included in the 2016 WHO classification system and is associated with pediatric and adult DIPG, gangliogliomas, and pilocytic astrocytomas.
MGMT Methylation
The MGMT gene is located on chromosome 10q26 and has a CpG-rich island with 98 CpG sites encompassing most of the promoter and the first exon. It normally catalyzes the transfer of a methyl group from the O6 position of a guanine nucleotide to a cysteine residue at the 145th position.Reference von Deimling, Korshunov and Hartmann71 It is a DNA repair gene, which must be replenished by the cell after being used in DNA restoration.Reference Hegi, Diserens and Gorlia72
With the use of alkylating chemotherapeutic agents act by the binding of an alkyl group to the O6 – position of guanine. This induces DNA mismatching, double-strand breakage, and ultimately cellular apoptosis. High levels of MGMT in cancer cells make them more resistant to alkylating chemotherapy, resulting in treatment failure. Methylation of the cytosine in CpG sites increases its affinity to proteins that prevent binding of transcription factors, effectively silencing MGMT expression. The epigenetic silencing of MGMT gene diminishes the tumor cell’s DNA repair capacity, making it more susceptible to the cytotoxic effects of alkylating chemotherapeutic agents.Reference von Deimling, Korshunov and Hartmann71,Reference Hegi, Diserens and Gorlia72
About half of all glioblastomas have methylated promoter sequences to the MGMT gene.Reference Hegi, Diserens and Gorlia72 MGMT promoter methylation is associated with longer survival (21.7-month median survival versus 15.3 months for those without methylation) and better responses to chemotherapy with alkylating agents.Reference Hegi, Diserens and Gorlia72,Reference Esteller, Garcia-Foncillas and Andion73 Secondary GBMs and astrocytomas with MGMT methylation are more likely to have a TP53 mutation and IDH mutations compared to those without methylation.Reference Nakamura, Watanabe, Yonekawa, Kleihues and Ohgaki74
Use of DNA Methylation in Classification of Gliomas
Integrating molecular markers and histological diagnoses into the 2016 WHO CNS tumor classification has allowed for better subtyping of gliomas but does not account for intra-tumoral heterogeneity within the same subtypes.Reference Paul, Mondal, Patil and Somasundaram75,Reference Weller, Pfister, Wick, Hegi, Reifenberger and Stupp76 The advent of DNA methylation profiling of gliomas allows for further refinement in diagnoses and integrating testing.
Principles of DNA Methylation Profiling in Gliomas
DNA methylation profiling involves identifying molecular difference among tumor groups as well as normal tissues. A hallmark of carcinogenesis is the disruption of normal gene regulation that leads to a loss or gain of function. Cytosine methylation is a stable epigenetic modification that may play a role in gene silencing.Reference Ordway, Bedell and Citek77 Promoter CpG island (CGI) hypermethylation results in gene silencing, characteristic of gliomas, and is often seen with global DNA hypomethylation.Reference Masui, Mischel and Reifenberger40,Reference Noushmehr, Weisenberger and Diefes78 DNA methylation, histone modification, and nucleosome remodelling all play an integral role in heritable gene silencing. Changes in these processes can result in pathologic gene silencing found in cancer.Reference Jones and Baylin79
Toyota et al.Reference Toyota, Ahuja, Ohe-Toyota, Herman, Baylin and Issa80 initially proposed the concept of a distinct CpG island methylation phenotype (CIMP) which was found in a subtypes of colorectal cancer.Reference van den Bent43 CIMP has since been used in studies in an attempt to refine glioma classifications. These studies will be discussed in the succeeding paragraphs of this paper.
Aside from studies of glioblastomas, genome-wide methylation profiling was also done in oligodendroglial tumors, with the work of van den Bent et al. showing CGI hypermethylation phenotype confers a better overall survival compared to unmethylated subgroup.Reference van den Bent and Gravendeel81
Noushmehr and colleagues had characterized a distinct subset of gliomas based on the presence of a G-CIMP using DNA methylation testing. Proneural subtype of GBM was seen to have a highly enriched G-CIMP cluster, while mesenchymal and classical subtypes’ G-CIMP clusters were found to be moderately enriched. Patients with G-CIMP-positive proneural tumors were noted to have a significantly younger age at onset and better survival compared to G-CIMP-negative proneural GBMs and other non-proneural GBM subtypes. IDH-1 mutation was seen to be tightly associated with G-CIMP positivity across all morphological glioma grades. G-CIMP positivity was observed to confer an improved survival outcome in these types of tumors. Independent of the IDH1 mutation status, G-CIMP has been shown to confer a favorable prognosis.Reference Noushmehr, Weisenberger and Diefes78,Reference Ceccarelli, Barthel and Malta82 The subtyping was studied further by Ceccarelli et al.Reference Ceccarelli, Barthel and Malta82 in 2016 when they concluded that IDH-mutant gliomas may be divided into three subgroups, namely: (1) the Codel group, which is composed of IDH-mutated, 1p/19q-codeleted low-grade gliomas; (2) the G-CIMP-low group, which included IDH-mutated non-1p/19q-codeleted gliomas that have low levels of DNA methylation; and (3) the G-CIMP-high group, including IDH-mutated 1p/19q non-codeleted gliomas with high levels of DNA methylation. They found that the G-CIMP-low group had a worse survival when compared to the 1p/19q-codeleted group and the G-CIMP-high group (log-rank p-value 3.9 × 10−10).Reference Ceccarelli, Barthel and Malta82 This showed that G-CIMP positivity conferred better survival outcomes independent of IDH mutation status.
Further molecular subtyping by Kloosterhof et al. divided gliomas into three groups based on their 1p/19q codeletion status: the “C-” subtype (CIMP– tumors, most GBMs and all samples of pilocytic astrocytoma), the “C+1p19q” (CIMP+, most AOs and a few GBMs), and “C+wt” (CIMP+, histologically diverse, mostly AA).Reference Kloosterhof, de Rooi and Kros83 Median survival for the C-, C+wt, and C+1p19q subtypes were 1.18, 2.62, and 5.00 years (14.16, 31.44, and 60 months), respectively. The sample size of pilocytic astrocytomas was too small to identify a separate subgroup. This study demonstrated that these three subtypes of gliomas had significant prognostic factors based on their characteristic methylation profiling.
CIMP on recurrence
The possibility of the G-CIMP-high group serving as a predecessor of the G-CIMP-low tumor type was investigated by Ceccarelli et al. by comparing the DNA methylation profiles of 10 IDH-mutated, non-1p/19 q-codeleted low-grade gliomas and GBMs with the TCGA cohort.Reference Ceccarelli, Barthel and Malta82 A clear (4 out of 10 cases) or partial (remaining 6 of 10 cases) demethylation pattern was noted after disease recurrence supporting the idea of a possible progression from a G-CIMP-high to a G-CIMP-low phenotype upon disease recurrence.Reference Ceccarelli, Barthel and Malta82 De Souza et al.Reference de Souza, Sabedot and Malta84 followed this up with their 2018 study where they observed that while most IDH-mutant LGG G-CIMP-high tumor patients retain their epimethyl phenotype on recurrence, there is a small proportion (17%) that underwent progression to a G-CIMP-low phenotype on relapse. A distinct subgroup with G-CIMP-intermediate profiles was characterized by a modest degree of DNA methylation changes that trends toward G-CIMP-low subtype suggesting that this group of tumors may be an early-stage transition from a G-CIMP-high tumor to a G-CIMP-low tumor. The group had also found a set of predictive biomarkers signatures which are composed of seven hypomethylated CpG sites in initially G-CIMP-high tumors that progressed to GBMs on recurrence. They were also able to note that 95% of tumors previously classified as IDH-mutant non-1p/19q-codeleted G-CIMP-low tumors belong to this “risk group” that was prone to progress to a G-CIMP-low IDH-wildtype GBM-like phenotype on recurrence.Reference de Souza, Sabedot and Malta84 We made a summary of the subgroups proposed by these studies in Figure 1. Of note, this figure shows grouping by DNA methylation studies as well as by the current (2016) WHO classification criteria in such a way that there is overlap with methylation and histological diagnoses.
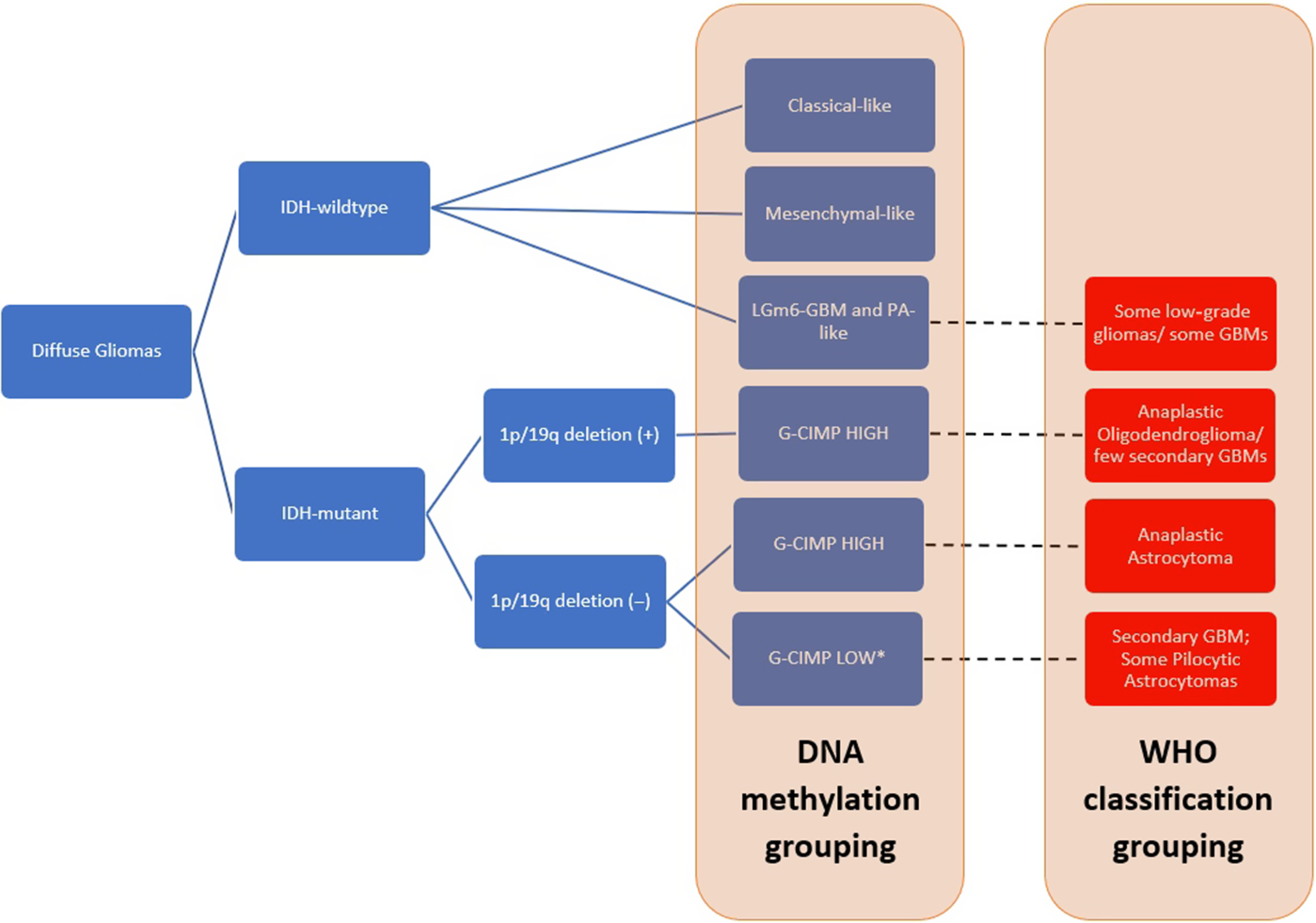
Figure 1: Summary of DNA methylation grouping based on the studies by de Souza et al.,Reference de Souza, Sabedot and Malta84 Ceccarelli et al.,Reference Ceccarelli, Barthel and Malta82 and Kloosterhof et al.Reference Kloosterhof, de Rooi and Kros83 as well 2016 WHO classification grouping. The diagram also shows that there is some overlap of histological groups when classified via DNA methylation testing. Red boxes represent the tumor samples classified based on WHO classification criteria, showing overlaps in the tumor grouping.
GBM Molecular Subtypes by DNA Profiling
With the advent of genome-wide DNA profiling, an increasing number of molecular subtypes are emerging. Phillips et al.Reference Phillips, Kharbanda and Chen85 were the first to coin the terms proneural, proliferative and mesenchymal to divide high-grade astrocytomas (AAs and glioblastomas) into molecular subclasses. They were able to observe that there is a mutually exclusive molecular markers expression pattern in the proneural and mesenchymal subgroups. The proliferative signature is less exclusive compared to the other two subgroups, but the gain of the PIK3R3 locus on chromosome 1 appears to be a unique alteration in this type of tumor. Cases segregated into the proneural subclass were found to be younger compared to mesenchymal or proliferative subclasses. Median survival was noted to be longer in the proneural subclass (174.5 weeks) compared to either proliferative (60.5 weeks) or mesenchymal subtypes (65.0 weeks) (Table 2).
Table 2: Distinguishing genetic alterations and corresponding median survival in patients with various GBM molecular subtypes

While a mixture in Grade III and IV gliomas was found in the proneural subclass, Grade IV gliomas predominated the mesenchymal and proliferative groups. The presence of some cases of Grade IV tumors in the proneural subclass may serve as evidence that molecular classification might prove to be superior to histological grading in prognostication.Reference Phillips, Kharbanda and Chen85
The GBMs sorted into the poorest prognostic molecular subgroups have a significantly shorter median survival compared to the ones in the most favorable molecular clusters (0.70 versus 2.05 years; (8.04 versus 24.6 months) p = 0.0024).Reference Gravendeel, Kouwenhoven and Gevaert86 Interestingly, when the different signatures previously identified by Phillips et al. were segregated into specific molecular clusters used by Gravendeel et al., one of the best prognostic clusters of Gravendeel is classified as poor prognostic group by Phillips.Reference Gravendeel, Kouwenhoven and Gevaert86 This discrepancy supports that while molecular profiling might be a promising alternative to histologic profiling, refinement is needed.
By integrating data from The Cancer Genome Atlas (TCGA), Verhaak et al. were able to subdivide GBM into four distinct subtypes based on patterns of gene expression, namely proneural, neural, classical, and mesenchymal. Each group differs in terms of genomic signature, age of the patient population, overall survival, and response to therapy (Table 3).Reference Verhaak, Hoadley and Purdom36
Table 3: Frequency of common mutations in four molecular subtypes as identified by Verhaak et al.Reference Taal, van der Rijt and Dinjens42 with frequencies

* p-Values at 0.01 level.
Examination of responses to aggressive treatment per subtype showed that there is a significant reduction in mortality in the mesenchymal and classical subgroups. Aggressive treatment was defined in the study as concurrent chemoradiotherapy or more than three cycles of chemotherapy. Although a noted trend toward longer survival was found in the proneural subgroup compared to other types of GBM, it was found not to improve with aggressive treatments.Reference Verhaak, Hoadley and Purdom36 In the analysis of the Rembrandt dataset by Guan et al.,Reference Guan, Vengoechea and Zhang87 the proneural subtype was noted to be more prevalent in lower grade gliomas compared to GBMs. Though further research would be beneficial, these studies indicate a potential use for integrating methylation studies into glioma classification.
Future Directions
The advent of molecular testing has led to increased knowledge of glioma subgroups, each with its characteristic molecular associations and clinical features. More recently, multiplatform and machine-learning strategies to classify gliomas have showed promising results. For example, through multiplatform comprehensive analysis (DNA methylation, mRNA expression, microRNA expression, and long noncoding RNA expression), Mao et al. were able to identify differential expression in IDH-mutated versus wildtype astrocytic tumors as well as four signatures in the calcium signalling pathway of prognostic significance.Reference Mao, Yin and Zhang88 Similar comprehensive analyses, though extensive, could allow for improved tumor classification beyond the 2016 WHO criteria.Reference Mao, Yin and Zhang88 As molecular testing in gliomas is performed more routinely globally and as more is understood about the clinical significance of these tests, opportunities for targeted therapies will increase. In the era of personalized cancer treatments, knowledge of unique molecular characteristics which are specific to various types of gliomas can provide molecular targets for future drug development and refinement of current agents available.
In the future, the ability to reliably predict and prognosticate clinical responses and survival in patients with varying grades and classes of gliomas based on detailed molecular testing would be beneficial for clinical practice. It will not only allow for more informed decision-making for patients with this terminal disease, but also allow for health care professionals to tailor personalized treatment strategies to optimize patient outcomes.
Conclusion
The emergence of in-depth molecular profiling has significantly changed the field of neuro-oncology in recent years. Since the release of the 2016 WHO classification of CNS tumors, further information on classification, clinical outcomes, and potential changes to management of diffuse gliomas has been achieved. This paper aimed to summarize key knowledge of important glioma biomarkers to date, updates provided by the cIMPACT-NOW consortium, and important studies on DNA methylation testing in diffuse gliomas. As further studies on comprehensive molecular testing and its use in glioma classification are available, the landscape of glioma classification will continue to evolve to reflect clinically relevant tumor classes.
Conflict of interest
There are no disclosures for any of the above authors.
Statement of authorship
All persons meeting authorship criteria have been included above, and all authors certify that they have participated sufficiently. Each other certifies that the material or similar material has not been submitted or published in any other publications and has only been submitted to the Canadian Journal of Neurological Sciences.
Conception or design of the work: WM.
Data collection, analysis, and interpretation: NK, RO, SB.
Drafting the article: NK, RO, SB.
Critical revision of the article: WM.
Final approval of the version to be published: NK, RO, SB, WM.




