



Vanishing white matter disease (VWMD) or childhood ataxia with central hypomyelination (CACH) is a common leukodystrophy characterised by autosomal recessive inheritance, progressive course and episodic deterioration provoked by stressors. Reference van der Knaap, Pronk and Scheper1 It is caused by pathogenic mutations in genes encoding the eukaryotic translation initiation factor complex (eIF2B1-5) subunits that exert their action via the guanine nucleotide exchange factor (GEF) activity. Most of the patients are from the Caucasian population from Europe and North America and only 9% are from Asia and Africa. Reference Hamilton, van der Lei and Vermeulen2 In all, 87% become symptomatic by 18 years of age and adult-onset VWMD is distinctly uncommon. Reference Hamilton, van der Lei and Vermeulen2
We describe two adult subjects from India with VWMD and eIF2B variations. The eIF2B variations were detected by clinical exome sequencing (Illumina platform). The actual sequencing, library preparation, and bioinformatics were outsourced to a private laboratory. No variations in genes implicated in other leukodystrophies were identified. Disability scoring was based on a 5-point scale adapted from Ohlenbusch et al. Reference Ohlenbusch, Henneke and Brockmann3 This study was approved by the Institute Ethics Committee (NIMHANS/105th IEC/2016, dated 18th July 2016). The clinical and radiological features and genetic variations are summarised in Table 1.
Table 1: Clinical characteristics and genetic variations in adults with vanishing white matter disease
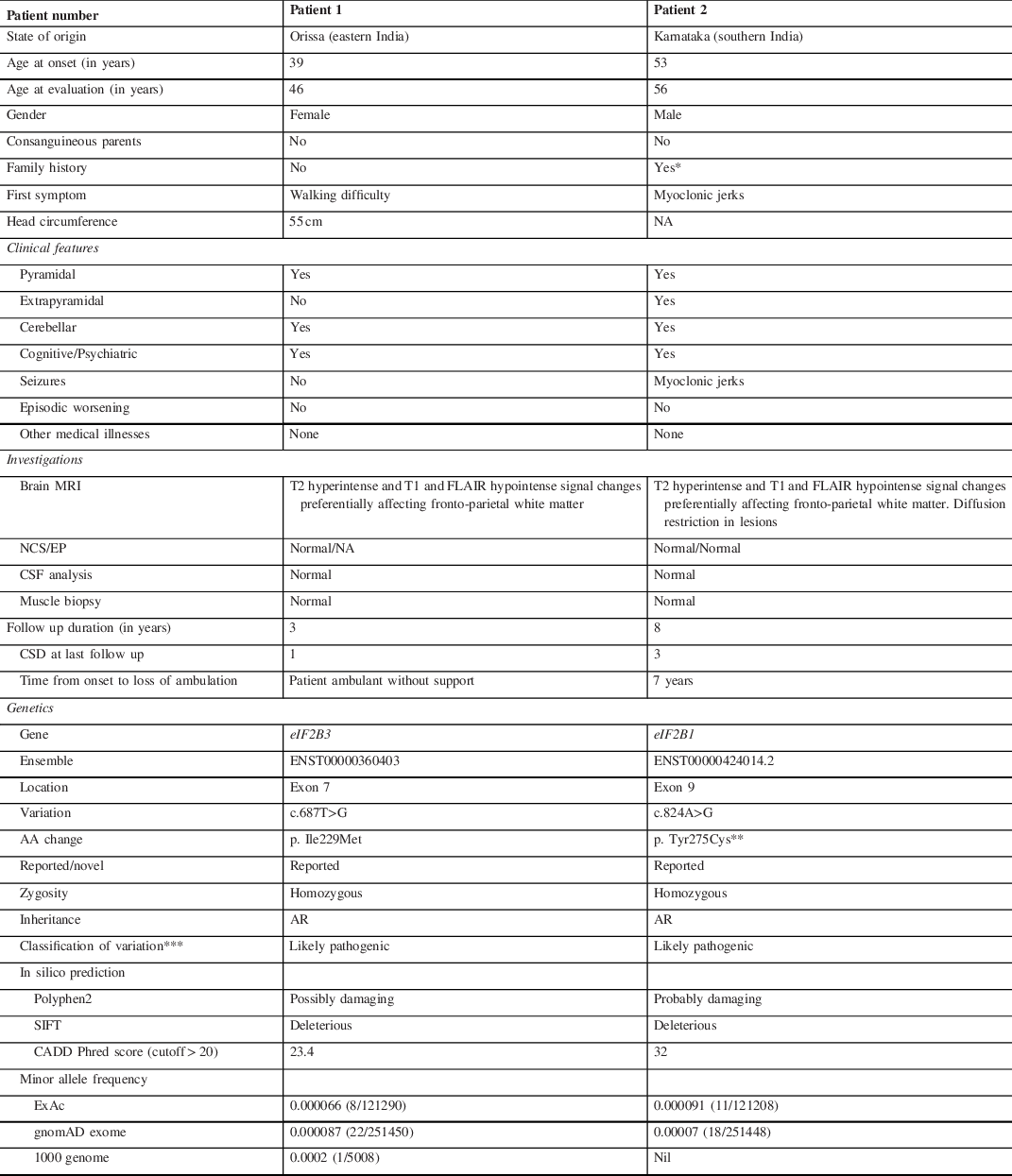
A: adenine; CADD: Combined Annotation Dependent Depletion; CSD: Clinical score of disability (by Ohlenbusch et al., 2005) – 0 = no functional handicap or neurological signs; 1 = mild coordination or gait difficulties not requiring assistance; 2 = moderate learning or neurologic abnormalities requiring support or intervention in a few areas, able to walk with assistance; 3 = severe learning or neurologic abnormalities requiring support in many areas, wheelchair-bound; 4 = requiring constant supervision, confined to bed, loss of cognitive abilities; 5 = death; CSF: cerebrospinal fluid; Cys: cysteine, G: guanine; EP: evoked potentials; Ile: isoleucine; LRT: Likelihood Ratio Test; Met: methionine; MRI: magnetic resonance imaging; NA: data not available; NCS: nerve conduction study; SIFT: Sorting Intolerant From Tolerant; T: thymine; Tyr: tyrosine.
* Mother of patient 2 had a neurological illness characterised by cognitive impairment, progressive motor disability and seizures but she was not evaluated for these complaints.
** This variant has been reported in the ClinVar (VCV000217281) by two submitters.
*** Classification was based on the American College of Medical Genetics and Genomics (ACMG) guidelines.
Patient 1 developed progressive walking difficulty from 39 years of age and tingling sensation in her lower limbs from 41 years of age. She underwent laminectomy for prolapsed lumbar intervertebral disc after which her sensory symptoms improved. She developed forgetfulness, apathy, irritability, delusions of infidelity and insomnia from 43 years of age. She was evaluated at our centre at 46 years of age. Examination revealed impaired episodic memory and construction ability. She had bi-pyramidal signs, impaired joint position sense, finger–nose–finger in-coordination and spastic gait. Brain MRI showed periventricular hyperintensities with rarefaction (Figure 1). Clinical exome sequencing revealed a homozygous missense variation [c.687T>G, (p. Ile229Met; ENST00000360403)] in the eIF2B3 gene, which has been reported previously in VWMD. Reference Pronk, van Kollenburg, Scheper and van der Knaap4 The reference codon is conserved across mammals. The variant was predicted to be deleterious by Polyphen-2, Sorting Intolerant From Tolerant (SIFT), and Combined Annotation Dependent Depletion (CADD) tools (Table 1). At the time of last follow-up at 49 years of age, the patient continued to be ambulant.

Figure 1: (A–E) and (F–J): Brain MRI of patient 1 and patient 2 respectively, showing white matter signal changes, predominantly involving the fronto-parietal region, which are hyperintense on T2W sequence, hyperintense with areas of cavitation on FLAIR sequence and hypointense on T1W sequence. (K–O): Brain MRI of patient 2, performed 7 years after first imaging shows an increase in cavitation and appearance of patchy diffusion restricted lesions.
Patient 2 developed myoclonic jerks at 53 years of age. From 56 years of age, he developed imbalance while walking, slowness, urinary incontinence, forgetfulness, and altered personality. He was first evaluated at our centre at 56 years of age. Examination revealed impaired executive function and verbal and visual memory. He had mask-like facies, bradykinesia, postural instability, bi-pyramidal signs, and finger–nose–finger incoordination. He walked independently with slow and short steps. Brain MRI showed periventricular hyperintensities with rarefaction (Figure 1). He was treated symptomatically with sodium valproate, levetiracetam, and clonazepam for myoclonic jerks and levodopa-carbidopa and amantadine for extrapyramidal features. He worsened and became wheelchair-bound at 60 years. Brain MRI at 63 years revealed pronounced cavitation (Figure 1). Clinical exome sequencing revealed a homozygous missense variation [c.824A>G (p. Tyr275Cys; ENST00000424014.2)] in eIF2B1 gene, which has been reported in ClinVar (VCV000217281) in patients with VWMD. Reference Maletkovic, Schiffmann and Gorospe5 The reference codon is conserved across species, and in-silico analysis by Polyphen-2, SIFT, and CADD predicted the variant to be damaging (Table 1). He was followed up till 65 years of age; he was dependent for all activities of daily living.
Till date, only a few patients of genetically established VWMD have been reported from India and none are adults. We previously reported five children with eIF2B variations. Reference Parayil Sankaran, Nagappa and Chiplunkar6 Based on the onset age, VWMD can be categorised into antenatal, early infantile (<1 year), late infantile (1 to <2 years), early juvenile (2 to <4 years), juvenile (4 to <8 years), late juvenile/adolescent (8 to <18 years) and adult (≥18 years) onset. Reference Hamilton, van der Lei and Vermeulen2 Hamilton et al., reported that age at onset was a key determinant for disease progression and survival: patients with onset before 4 years of age had rapid progression, earlier loss of ambulation and higher mortality as compared with those with later onset. Reference Hamilton, van der Lei and Vermeulen2 Labauge et al., reported that even in adult-onset VWMD, nearly 80% lost independent ambulation. Reference Labauge, Horzinski and Ayrignac7 In our cohort, patient 2 became wheelchair bound at last follow-up whereas patient 1 remained ambulant. Thus, factors that determine progression in adult-onset VWMD such as epigenetics, modifier genes and environmental factors remain to be understood. Female predilection has been reported whereas males have more severe disease. Reference Labauge, Horzinski and Ayrignac7
The commonest manifestation of VWMD is a spastic-ataxic syndrome, Reference Hamilton, van der Lei and Vermeulen2,Reference Labauge, Horzinski and Ayrignac7 as was noted in the present study as well as in children reported by us earlier. Reference Parayil Sankaran, Nagappa and Chiplunkar6 Psychiatric and cognitive symptoms may precede other symptoms in adults. Reference van der Knaap, Pronk and Scheper1 Both the patients in the present report developed cognitive and/or psychiatric symptoms during the disease course, whereas these were not noted in our cohort of children. Reference Parayil Sankaran, Nagappa and Chiplunkar6
Seizures are common, usually occur 1 to 2 years after onset, are usually focal, and may indicate a worse prognosis. Reference Hamilton, van der Lei and Vermeulen2,Reference Labauge, Horzinski and Ayrignac7 Refractory epilepsy is not a feature of adult-onset VWMD. Reference Hamilton, van der Lei and Vermeulen2 Patient 2 had myoclonic jerks as the first symptom. Other neurological features described in VWMD are mild intellectual disability, optic atrophy, chorea, tremor, torticollis, and hemiplegic migraine-like episodes. Reference van der Knaap, Pronk and Scheper1 Patient 2 in the present study had features of parkinsonism. Patient 1 had neither seizures nor extrapyramidal features.
Ovaries are a common site of extra-neural involvement leading to the terminology ‘ovario-leukodystrophy’. Primary/secondary amenorrhoea and rarely menometrorrhagia may occur and these may precede neurological symptoms. Our patient (patient 1) did not have evidence of ovarian failure. Several other non-neurological manifestations have been described Reference van der Knaap, Pronk and Scheper1 but none were observed in our cohort.
A distinctive feature of VWMD is episodic neurological worsening triggered by fever, trivial head injury, infections, fear, stress, vaccination, and pregnancy. Reference van der Knaap, Pronk and Scheper1 Patients may recover from this temporary worsening but not necessarily completely and occasionally these episodes may be fatal. This episodic pattern may mimic the relapsing–remitting course of acquired immune-mediated diseases such as multiple sclerosis. None of the adults in the present study had episodic worsening. Other studies also reported that episodic worsening is less common in adults as compared with children (Supplementary tables 1 and 2).
Both patients in the present study had similar MRI findings characterised by hyperintensity of white matter (WM) on T2 and hypointensity on T1 and FLAIR. Fronto-parietal WM is preferentially affected, whereas the temporal WM, optic nerves, outer edge of the corpus callosum, anterior commissure and internal capsule are relatively spared. Reference van der Knaap, Pronk and Scheper1 Within the abnormal WM, radiating stripes of hyperintensity are noted which correspond to the spared areas around the blood vessels. Reference van der Knaap, Pronk and Scheper1 Extensive WM changes have been reported even in pre-symptomatic or mildly disabled patients. Reference van der Knaap, Pronk and Scheper1,Reference Labauge, Horzinski and Ayrignac7 MRI abnormalities progress over time and in advanced stages the WM ‘vanishes’ leaving behind only the ventricular margins and the overlying cortex. Patient 2 had increased cavitation in serial MRIs seven years apart (Figure 1). Other MRI findings have been reported. Hyperintensities may be seen in the brainstem, particularly in the central tegmental tracts. Reference van der Knaap, Pronk and Scheper1 Cerebellar involvement has been reported in childhood and adult-onset VWMD and more often consists of atrophy rather than abnormal signal alterations. Reference van der Knaap, Pronk and Scheper1,Reference Labauge, Horzinski and Ayrignac7 Cranial nerve enhancement and chronic subdural haematoma have been reported in isolated cases with childhood/infantile-onset VWMD. Reference Eluvathingal Muttikkal, Montealegre and Matsumoto8,Reference Güngör, Güngör and Çakmaklı9 Hyperintensities of the globus pallidi and thalami have been noted uncommonly in children Reference van der Knaap, Pronk and Scheper1 but not in adult-onset VWMD. Reference Labauge, Horzinski and Ayrignac7 These changes were not noted in the present study.
The WM changes were initially considered to be due to hypomyelination, hence the term CACH. Subsequent studies showed evidence of rarefaction and cystic degeneration of WM without inflammation, which matched the MRI changes. Microscopically, astrocytes and oligodendrocytes are preferentially affected and VWMD has been classified as an ‘astrocytopathy’. During cellular stress, mRNA translation and protein synthesis are inhibited, a process referred to as the unfolded protein response (UPR). Reduced eIF2B activity in VWMD leads to an increase in UPR and increased denatured proteins in the endoplasmic reticulum. In VWMD, the cells appear to be hyperreactive to stress. Reference van der Knaap, Pronk and Scheper1 Mutations in the eIF2B5 are reported to be common among Caucasians, whereas Asian studies found a relatively lower frequency. Reference van der Knaap, Pronk and Scheper1 None of our patients, including the children previously reported by us, had mutations in eIF2B5. Reference Parayil Sankaran, Nagappa and Chiplunkar6 Certain mutations have been shown to predict the clinical course. Majority are missense mutations. Nonsense mutations are rare, are always heterozygous and occur in association with another missense mutation, because their presence in homozygous states is not compatible with survival. No precise correlation between the mutated subunit or domain with the phenotype has been established. The p. Arg113His and p. Arg195His in the C-terminal domain of the eIF2B5 are associated with mild and severe phenotypes, respectively. Variations in the non-conserved regions may contribute to a milder phenotype and variations in highly conserved regions to more severe phenotypes. The type of amino acid substitution such as the one that causes a significant change in the 3-D structure of the protein may correlate with a severe phenotype. Reference Fogli and Boespflug-Tanguy10
This study describes adults with genetically established VWMD from India emphasising the clinical course and serial radiological abnormalities. Clinicians should suspect this disease in adults based on the MRI findings and confirm the diagnosis by demonstrating eIF2B mutations as it has implications for genetic counselling and prognostication. It would be worth analysing a larger cohort of patients with genetically established adult-onset VWMD from multiple centres across India and Asia for the natural course and survival.
Disclosures
None of the authors have any disclosures.
Ethics Approval
The study was approved by the Institute Ethics Committee of the National Institute of Mental Health and Neurosciences (NIMHANS), Bangalore, India (No: NIMHANS/105th IEC/2016, dated 18th July 2016). The study was performed in accordance with the ethical standards as laid down in the Declaration of Helsinki.
Statement of Authorship
Study concept and design: Nagappa M and Shivaram S. Study supervision: Nagappa M, Sinha S, Bindu PS and Taly AB. Analysis and interpretation of genetic data: Shivaram S, Nagappa M, Seshagiri DV, Sinha S, Bindu PS, Taly AB and Govindaraj P. Clinical data collection and MRI interpretation: Shivaram S, Nagappa M, Seshagiri DV and Saini J.
Supplementary Material
To view supplementary material for this article, please visit https://doi.org/10.1017/cjn.2021.202.




