


A male of Western European descent and living in Canada began experiencing progressive insomnia, asymptomatic tachycardia and diaphoresis at the age of 46. Twenty-two months prior to the onset of insomnia, he was diagnosed with diabetes, and 16 months prior hypertension (Figure 1A).
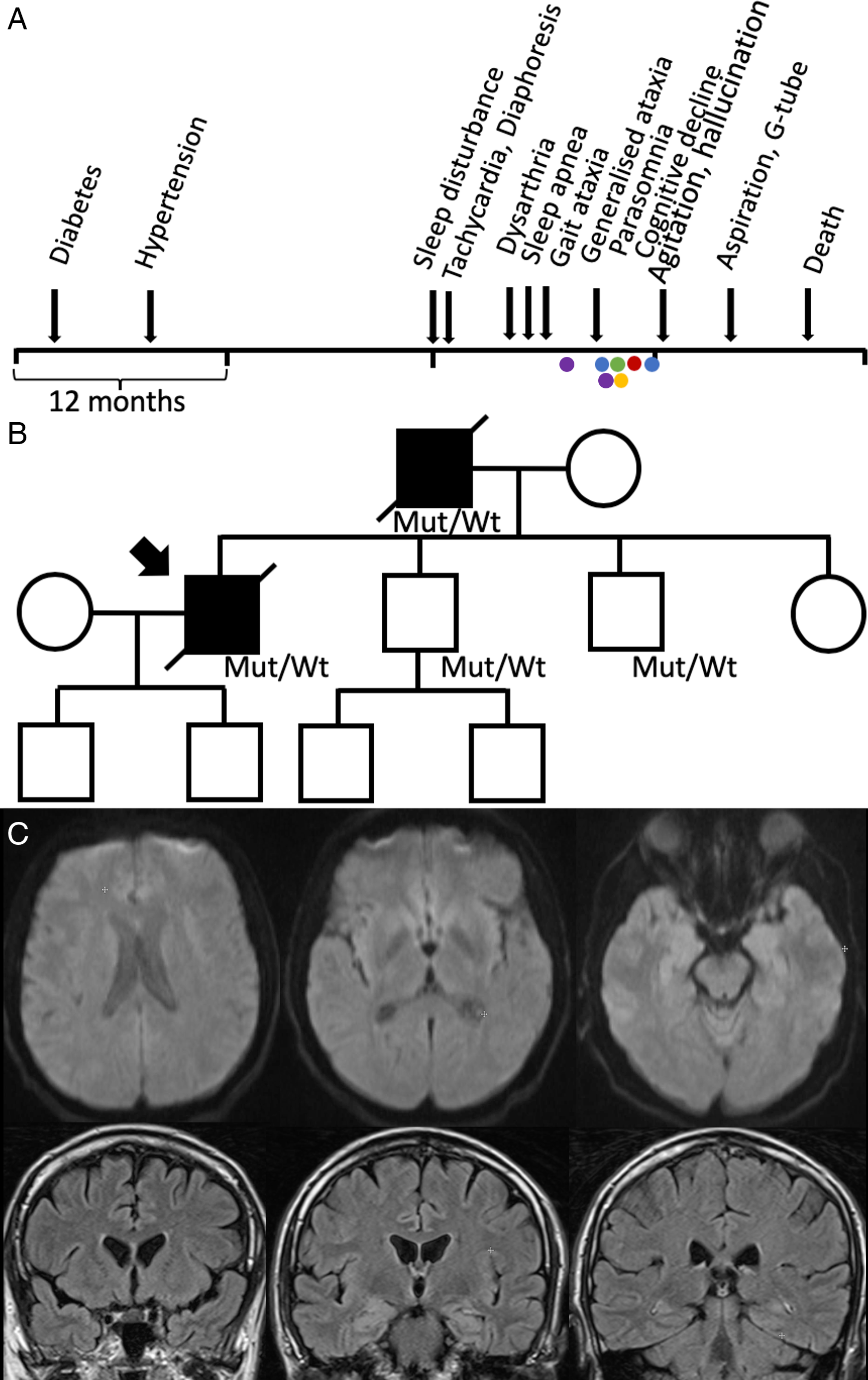
Figure 1: (A) Clinical timeline. Electroencephalogram (blue), tilt table test (green), echocardiogram (red), polysomnogram (yellow) and MRI brain (purple). (B) Pedigree. The patient’s father had a similar presentation and died at age 48. His brain tissue pathology was consistent with FFI; therefore, we assumed he likely also had the same mutation. The mutation was confirmed in his two brothers; however, they remain asymptomatic. (C) The second MRI head performed with diffusion weighted imaging (top) and FLAIR post gadolinium (bottom), showing no acute pathology.

Figure 2: (A) Polysomnography (30 second epoch) showing wakeful appearing EEG; however, also shows roving eye movements, typically associated with REM sleep. Patient appeared asleep during recording. (B) Polysomnography recording during wakefulness showing central apnea (highlighted in pink).
Four months following the onset of insomnia, he experienced intermittent dysarthria. One month later sleep apnea appeared, and progressive gait ataxia emerged the subsequent month. He was admitted to our hospital 2 months later for progressive dysarthria, dysphagia and gait imbalance. He was noted to have tachycardia (heart rate 110–125) which was determined by cardiology to be non-structural, though no clear aetiology was identified.
The month following his discharge he reported cognitive decline, parasomnias and worsening sleep apnea. He noted diplopia and his examination revealed impaired saccades, saccadic intrusions and bilateral end-gaze nystagmus. His dysarthria worsened and was associated with pressured and unintelligible speech and difficulty chewing. He became ataxic, his gait declined, and he became wheelchair-bound. There was no sensory or strength impairment. He was admitted to hospital and given intravenous immunoglobulin, without improvement.
In the next 3 months, his cognition further declined, and he could no longer recognise his family members. He became more active during sleep, with recurrent vivid dreams, rapid eye movement sleep behaviour disorder and periods of inappropriately falling asleep during the day. He could not sustain eccentric gaze. He began aspirating. He had mild hypercapnia PCO2 48–50 mmHg. He underwent polysomnography which demonstrated severe central sleep apnea (CSA) and status dissociatus (Figure 2). He was started on BiPAP.
His A1C was 7.3 and triglyceride was 2.65 on his first admission. Otherwise, haematologic, metabolic, inflammatory, infectious and paraneoplastic investigations were unremarkable. Spinocerebellar ataxia panel, fragile X-associated tremor/ataxia syndrome, Huntington’s, acetylcholine receptor antibody, antigliadin antibody, tissue transglutaminase IgA and genetic testing for Nieman–Pick were negative. Cerebrospinal fluid showed no pleocytosis and negative 14-3-3. Real-time quaking-induced conversion was not conducted. His urine metanephrines showed a mild elevation of normetanephrines (3.9 umol/day, normal range: <3.4 umol/day) and metanephrines (1.6 umol/day, normal range: <1.5 umol/day).
Two magnetic resonance imagings of the brain with diffusion weighted imaging and FLAIR were performed (second with gadolinium), which were nondiagnostic. Electroencephalogram initially was normal and subsequently showed grade II dysrhythmia. Electromyography was normal. Echocardiogram was normal. Computed tomography neck, chest, abdominal and pelvis and scrotum ultrasound for malignancy screen were negative. Single-photon emission computed tomography head was negative. Tilt table test was negative. Videofluoroscopic swallow study showed oropharyngeal dysphagia.
The next month, he was re-admitted with worsening confusion, agitation, self-mutilation, hallucinations and ataxia. All activities of daily living were impaired. Four months later, he had aspiration pneumonia, and a G-tube was inserted. He had two recurrent episodes of aspiration pneumonia in the subsequent months, and he passed away 20 months following the onset of insomnia. His genetic test was positive for PRNP D178N/129M heterozygous at codon 129, consistent with a diagnosis of Fatal Familial Insomnia (FFI).
His autopsy reported focal subtle microspongiosis of the deeper cortical layers of the cingulate gyrus and the basotemporal cortex. Dorso-medial thalamus, hypothalamic structures and inferior olivary nucleus of the medulla showed subtotal neuron loss with extensive gliosis. There was moderate cerebellum Purkinje cell loss, with formation of axonal torpedoes and some Bergmann gliosis.
Family history was significant for a father with progressive sleep disturbance and death at the age of 47. The father had dysautonomia, visual and auditory hallucinations, cognitive decline and sleep apnea. His brain tissue pathology was also consistent with FFI. His father’s brother was murdered in his 40s, and sister died of heart disease in her 80s. He is not in touch with their children. His father’s parents passed away in their 60s and 70s. His two brothers are 42 and 45, both gene carriers, but presently asymptomatic. His sister is 49 and also healthy. He has two sons (14 and 17 years old), and his younger brother has two sons, all healthy and not genetically tested (Figure 1B).
FFI is an autosomal dominant prion disease, resulting from a missense mutation on the prion protein gene PRNP D178N/129M. Reference Baldelli and Provini1 FFI is characterised by agrypnia excitata, involves multiple systems with considerable clinical and pathological variability. Sleep disorders generally start first, while dysautonomia, movement disorders, cognition and other prion disease symptoms follows.
Dysautonomia is a key feature of FFI and may be secondary to early selective dorso-medial and anteroventral thalamic degeneration resulting in thalamo-limbic interruption. Reference Cortelli, Perani and Montagna2 The frequency and timing of onset of hypertension and tachycardia vary between reports and may accompany early insomnia in some cases. Reference Baldelli and Provini1,Reference Wu, Zhan and Huang3,Reference Rodriguez-Porcel, Ciarlariello and Dwivedi4
Although various endocrine changes may be seen in early FFI, diabetes is generally not associated with FFI. Reference Baldelli and Provini1 One study showed an FFI patient without a history of diabetes had marked insulin resistance, although it is unclear when the resistance started. Reference Benedini, Cortelli and Caumo5 Lu reported a case of FFI with long standing hypertension, and a new incidental A1C finding of 6.5% (6.0% 2 months ago) and a co-diagnosis of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Reference Lu, Pan and Peng6
Interestingly, in our case, diabetes and hypertension preceded sleep disturbance, and tachycardia was also initially documented around that time; however, it was in the context of hyperglycaemia. Subsequently, tachycardia was reported shortly after the onset of sleep impairment. While echocardiogram and table tilt testing were negative.
Although the diabetes could be coincidental given that he has a family history of diabetes, the fact that diabetes and hypertension both developed within 22 months prior to the onset of sleep disturbance in a relatively young patient and that tachycardia and diaphoresis also occurred shortly after suggest that this maybe associated with underlying FFI pathology.
It is important to assess endocrine and autonomic function in FFI. The variability in FFI-related dysautonomia may in part be due to inconsistent reporting. Reference Baldelli and Provini1 Furthermore, we know that not all FFI patients present with sleep disturbance as their initial symptom. In healthy carriers that are genetically confirmed with FFI, endocrine and autonomic impairments may be used as an early marker of clinical disease onset and therapeutic interventions.
Statement of Authorship
- RL:
-
Drafted the manuscript.
- MM:
-
Part of initial diagnosis of patient, manuscript edits.
- NB:
-
Participated in care of patient.
- DB:
-
Patient’s family physician.
- ST:
-
Patient’s neurologist.
- LL:
-
Patient’s neurologist, manuscript edits.


