


Nemaline myopathy is a clinically and genetically heterogenous disorder that may present from birth to late adulthood. Patients with nemaline myopathy exhibit weakness of proximal muscles (particularly neck flexors) and, in the case of younger children, facial, bulbar, and/or respiratory muscle weakness. Nemaline myopathy can be classified according to age of symptom onset and disease severity.Reference Ryan, Schnell, Strickland, Shield, Morgan and Iannanccone 1 It has been linked to nine genes: NEB, ACTA1, TPM2, TPM3, TNNT1, CFL2 and less commonly; KLH40, KLH41, and KBTBD13.
The classic features seen on muscle biopsy include sarcoplasmic rodlike structures on microscopic study using Gomori trichrome stain. Electron microscopy also classically demonstrates electron-dense nemaline bodies associated with sarcomeric Z-lines and less commonly intranuclear inclusions.
Skeletal muscle alpha-actin (ACTA1) is the principal actin isoform of skeletal muscle. It forms the thin filament of the sarcomeric unit and interacts most notably with myosin to generate muscle contraction. Mutations within ACTA1 are the second most common cause of nemaline myopathy (after NEB mutations) accounting for about 20%-30% of cases.Reference Laing, Dye, Wallgren-Pettersson, Richard, Monnier and Lillis 2 Most ACTA1 mutations are thought to be de novo, although autosomal dominant and recessive inheritance has been reported.Reference Laing, Dye, Wallgren-Pettersson, Richard, Monnier and Lillis 2
We provide a clinical summary of an infant who presented with failure to thrive and tachypnea and who was later diagnosed with a nemaline myopathy. We illustrate the value of MRI in this case at assisting with the selection of muscle to biopsy. Genetic testing subsequently confirmed a mutation in an ACTA1 gene. Clinical follow-up was favourable as with adequate nutrition and bilevel positive airway pressure ventilation support the patient demonstrated appropriate gross motor development at his most recent clinical follow-up at 21 months old.
Case
A 4-month-old boy presented to the emergency room for failure to thrive over the preceding 2 months. He was born at 37 weeks’ gestational age to a healthy mother. Pregnancy was unremarkable. Antenatal ultrasounds were reassuring with no polyhydramnios. Delivery was via a planned Caesarean section with no resuscitation required. His birth weight was 3.32 kg (75th %ile). He established bottle-feeding without difficulty and was discharged home. His mother reported a gradual increase in fatigue with feeding, poor weight gain, and rapid breathing over at least 2 months. Physical examination showed an alert infant with tachypnea, mild intercostal indrawing, and abdominal breathing. No dysmorphic features were apparent. His weight was 4.59 kg (below third %ile) and head circumference was 41.8 cm (50th %ile). Cardiorespiratory and abdominal examination was unremarkable. Cranial nerve examination noted only a slight decrease in facial expression. His eye closure was strong and no ptosis or extraocular eye movement abnormality was apparent. His muscle power and tone were decreased. When pulled from a supine-to-sitting position, he demonstrated near-complete head lag. He was moving his extremities well and demonstrated antigravity movements with his arms and legs. He would bring his hands to his mouth and would grasp objects held in front of him. His grip strength was normal. He was not able to roll. He appeared weaker when trying to pull away from the examiner; however, his motor examination did not reveal definite weakness affecting any particular muscle group. Deep tendon reflexes were decreased (1+), but could be elicited. No contractures, scoliosis, or scapular winging was noted. He required pediatric intensive care unit admission for mask continuous positive airway pressure, nasogastric feeding, and diagnostic workup.
Further neurodevelopment history noted prior gross motor delay. He could briefly keep his head in line with his body when sitting with support. He was making attempts to roll. When supine, he could bring both hands to his mouth, reach up for objects, and briefly hold a bottle. He was attentive, vocalizing, visually tracking, and initiating smiles. Family history noted that his father had multiple sclerosis. His mother and sister (11 years old) were well. There was no parental consanguinity. There was no neuromuscular, metabolic, or cardiorespiratory disease among extended family members.
He required continuous day and night positive airway pressure for chronic compensated respiratory acidosis. Cardiology evaluation and echocardiogram was normal. Respirology testing included chest radiograph and CT and revealed mild atelectasis but no intrinsic lung pathology. Thoracic ultrasound revealed bilateral decreased in diaphragmatic excursion. Electrodiagnostic testing revealed normal median, ulnar, peroneal, and sural sensory response as well as normal median, ulnar, peroneal, and tibial motor responses. Concentric needle electromyogram of the iliopsoas, vastus lateralis, gastrocnemius, and tibialis anterior were normal. Serum creatine kinase was 21 U/L (normal<150 U/L). Serum lactate, plasma amino acids, and MRI of the brain was normal. MRI of the proximal lower limbs noted the axial T2 fast spin echo (FSE) to show marked signal abnormality in the hamstring muscles (Figure 1A). Given the findings on MRI, a muscle biopsy of the right hamstring was performed. Gomori trichrome staining revealed abundant granular or rodlike structures within the sarcoplasm of several muscle fibers (Figure 1B). Electron microscopy revealed sarcoplasmic nemaline rods or rodlike structures, at times associated with the Z bands (Figure 1C), consistent with the diagnosis of nemaline rod myopathy. Genetic testing identified a mutation in ACTA1 (NM_001100.3:c.407T>C, p.Val136Ala). Sequencing of NEB, TNNT1, TPM2, TPM3, CFL2, and MTM1 revealed no mutations.
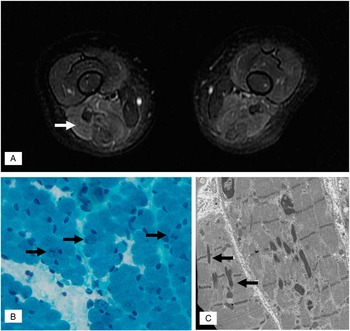
Figure 1 (A) MRI of the proximal leg muscles noted axial T2-weighted FSE revealed hyperintense signal in the posterior compartment of the thigh (white arrow) with relative sparing of the anterior compartment. (B) Gomori trichrome stain showing some fibers (black arrows) with sarcoplasmic dark stippling or rare elongated rods. (magnification: 40×). (C) Electron micrograph illustrating two longitudinally oriented skeletal muscle fibers containing several nemaline rods. Some small rods (black arrows) are clearly associated with Z bands (magnification: 12,000×).
He demonstrated good weight gain with nasogastric and later gastrostomy tube feeding. At his most recent clinical follow-up at 21 months old, he had discontinued using his gastrostomy tube and had been exclusively feeding by mouth for the prior 6 months. His weight was 11.2 kg (25th %ile). He had started crawling at 7 months old and pulling to stand at 12 months old. He walked independently at 14 months old. He was able to squat down to pick up a toy and rise back up with a negative Gowers sign. He did show persistent, mild facial diplegia and his mouth tended to droop open when relaxed. He continues to be gradually weaned off overnight bilevel positive airway pressure.
Genetic testing of his parents found his asymptomatic mother to also carry the ACTA1 mutation. A physical examination of the mother revealed no muscle weakness and there was no history in keeping with childhood weakness or failure to thrive. Maternal grandparents were tested and found not to carry the mutation consistent with a de novo mutation in the patient’s mother.
Discussion
Mutations in ACTA1 have been associated with mild-to-severe disease phenotype. Clinically, our patient demonstrated typical congenital nemaline myopathy. Based upon family history, he was initially suspected to have a sporadic mutation; however, subsequent testing of his parents identified his asymptomatic mother to possess the same ACTA1 mutation. Sequencing of the patient’s maternal grandparents confirmed this to be de novo in the mother. This mutation has been reported in another family that also demonstrated significant phenotypic variability among family members.Reference Agrawal, Strickland, Midgett, Morales, Newburger and Poulos 3 ACTA1 mutations have also been reported to demonstrate incomplete penetrance that has been explained by somatic mosaicism in a parent of a severely affected child.Reference Agrawal, Strickland, Midgett, Morales, Newburger and Poulos 3 The patient’s mother demonstrates either incomplete penetrance or somatic mosaicism given her normal neurological assessment and the de novo mutation detected in her lymphocytes. Definite evidence to support somatic mosaicism would require her to undergo a muscle biopsy demonstrating reduced or absent ACTA1 mutation in her muscle cells, which was not pursued in this case.
The differential diagnosis of infants presenting with facial and limb weakness with or without respiratory distress is broad and can include lesions throughout the neuroaxis, including disorders of: muscle (e.g. congenital myopathies, congenital muscular dystrophies, and metabolic myopathies including glycogen storage disease type II); neuromuscular junction (e.g. congenital myasthenic syndrome, transient myasthenia gravis); peripheral nerve (e.g. spinal muscular atrophy with respiratory distress syndrome, infantile Guillain-Barré syndrome); motor neuron disease (e.g. spinal muscular atrophy), spinal cord (e.g. infectious myelitis), or brainstem (e.g. syringobulbia or Möbius syndrome). Our patient did not demonstrate any clinical evidence for a central lesion and no electrophysiological evidence for neuropathy or neuronopathy. MRI of the muscle was particularly helpful at confirming involvement of this tissue.
MRI of the muscle was also key for guiding the selection of muscle that was biopsied in this patient. The identification of nemaline rods upon light microscopy facilitated a relatively rapid diagnosis of nemaline myopathy and directed further genetic testing. Unless otherwise specified, the quadriceps muscle biopsy is typically performed for infants at many centres given ease of access and the larger size of this muscle. In our patient, it is uncertain whether a quadriceps biopsy would have been as likely to have yielded evidence of nemaline rods given the relatively normal appearance of the quadriceps muscle on MRI relative to the hamstrings (Figure 1A). Recently, a child with nemaline myopathy resulting from a de novo ACTA1 mutation was reported to have undergone two nondiagnostic quadriceps muscle biopsies (at 8 months old and 8 years old). Each biopsy yielded nonspecific findings that included variable fibre diameter and marked type 1 fibre predominance. Only after an MRI-guided third biopsy of his deltoid muscle (at 19 years old) were nemaline rods identified.Reference Castiglioni, Cassandrini, Fattori, Bellacchio, D’Amico and Alvarez 4 MRI has been employed for many myopathies including nemaline, with some authors proposing that specific patterns of muscle involvement may offer predictive value for diagnosis and/or directing genetic tests.Reference Jungbluth, Sewry, Counsell, Allsop, Chattopadhyay and Mercuri 5 Nemaline myopathy resulting from NEB mutations has been reported to cause predominant distal leg atrophy, whereas ACTA1 has been linked to proximal leg muscle involvement.Reference Jungbluth, Sewry, Counsell, Allsop, Chattopadhyay and Mercuri 5 Our patient demonstrated involvement of proximal leg muscles but, unlike the prior report, he demonstrated predominantly posterior compartment (hamstring) involvement and not diffuse atrophy of the thigh. Our patient demonstrated normal MRI (axial T2WI FSE) of his gluteus maximus, gluteus medius, hip adductors, quadriceps, and gastrocnemius muscles. At this time, there are insufficient data to comment upon whether specific nemaline myopathy gene mutations give indeed rise to predictable patterns of muscle involvement. Despite this, we have found MRI of the muscle to be a valuable tool at assisting in the selection of which muscle to biopsy in patients with suspected myopathies.
Disclosures
None.

