


The long-chain PUFA (LC-PUFA) DHA is found in high concentrations in the retina and the brain. Pioneering work in the early 1990s demonstrated that addition of DHA, using fish oil supplements, to infant formulas of the day resulted in improved visual acuity compared with matched, unsupplemented formulas in both preterm(Reference Birch, Birch and Hoffman1, Reference Carlson, Werkman and Tolley2) and term infants(Reference Makrides, Neumann and Simmer3). However, some of these early studies suggested that infants fed the fish oil-supplemented formulas had lower weight and length gains than unsupplemented infants and that this was related to the reduction in arachidonic acid (AA) status caused by the addition of EPA and DHA found in the marine oils used in the studies(Reference Carlson, Werkman and Tolley2, Reference Carlson, Cooke and Werkman4). Although most subsequent trials have shown no negative effect of LC-PUFA supplementation on growth(Reference Makrides, Gibson and Udell5), they have tended to evaluate formulas containing DHA and AA with little or no EPA. While this mirrors the composition of most currently marketed formulas with LC-PUFA, there is an absence of systematic studies involving newborn infants to assess the potential impact of the AA–EPA balance in infant formulas. AA and EPA have important and often contrasting functions as precursors of eicosanoids and cyclo-oxygenase products, and modulate immune and inflammatory responses(Reference Kew, Mesa and Tricon6). Only two small studies from the same laboratory have assessed the effect of infant formulas supplemented with DHA and AA on immune markers, and showed some modulation in lymphocyte populations and cytokine production(Reference Field, Thomson and Van Aerde7, Reference Field, Van Aerde and Robinson8).
Breast-feeding promotes the colonisation of bifidobacteria and lactobacilli that inhibit the growth of pathogenic micro-organisms and compete with potentially pathogenic bacteria for nutrients and epithelial adhesion sites. The gastrointestinal flora may modulate mucosal physiology, barrier function and systemic immunological and inflammatory responses(Reference Agostoni, Axelsson and Braegger9–Reference Newburg12). It is with this rationale that bifidobacteria and other probiotic strains have been added to infant formulas. Studies show that probiotic supplementation favourably alters infant gut microbiota and some studies suggest that certain probiotic bacteria added after 5 months of age may protect against diarrhoea and reduce the onset of allergy(Reference Isolauri, Arvola and Sutas13–Reference Szajewska and Mrukowicz17). However, it should be noted that most trials assessing the safety and efficacy of probiotics in infant formulas have not included infants in the first 4–6 months of age who are more vulnerable and have a less developed immune system than their older counterparts(Reference Agostoni, Axelsson and Braegger9).
We evaluated an infant formula for term infants containing a probiotic, Bifidobacterium lactis, and the LC-PUFA DHA and AA. Although these components have been added commercially and are often accepted as safe(Reference Makrides, Gibson and Udell5, Reference Saavedra, Bauman and Oung16, Reference Kullen and Bettler18–Reference Simmer and Patole21), formulas containing both components have not yet been studied for safety in term infants. For this reason, the primary aim of the present study was to assess the safety of the probiotic-LC-PUFA combination by comparing the growth of term infants who were fed the experimental formula with that of infants fed a standard infant formula. Growth monitoring remains the best method to assess the well-being of preverbal children(Reference Simmer20, Reference Simmer and Patole21). As both probiotics and LC-PUFA have the potential capacity to alter immune function and there is a theoretical basis for synergistic action(Reference Das22), we also compared the antibody response of infants to vaccination, as well as how the two test formulas were tolerated. Antibody response to vaccination was chosen as the immune outcome because it represents a systemic response related to a clinically important outcome(Reference Hawkes, Gibson and Roberton23).
Methods
Participants
Mothers of infants who chose to exclusively formula-feed their infants were approached to enter the trial. Term ( ≥ 37 weeks) and newborn infants ( ≤ 10 d old) with birth weight between 2500 and 4500 g were enrolled. Infants were excluded from the study if: they had any congenital malformations or significant postnatal diseases; they were participating in another clinical trial; the parents were unwilling to have infants immunised according to the National Health and Medical Research Council (NHMRC) schedule(24) they were unlikely to attend follow-up visits.
Trial design
The present study was a prospective, double-blind, controlled, parallel-group, single-centre trial performed in Adelaide, Australia (Women's and Children's Hospital, Children, Youth and Women's Health Service (CYWHS)) between August 2003 and May 2005. The primary objective was to demonstrate bioequivalence of the experimental and control formulas based on equivalent weight gain. The secondary objectives were to assess the effects of the formula on immune response to immunisation and blood biochemistry, infant tolerance and morbidity.
Blinding of formulas was achieved by assigning two colour codes to each test formula. Sealed, opaque envelopes were prepared according to an independently generated randomisation schedule, stratified by sex. Upon enrolment, investigators gave the mothers the allocated formulas according to the coding in the concealed envelopes along with instructions on preparation. Infants were fed ad libitum their allocated formula starting from the day of enrolment until they were 212 ± 7 d old (7 months). Infants were allowed to take weaning foods starting from 4 months of age but they also had to be fed with at least 500 ml/d of their allocated formulas during this time. Follow-up home visits took place when infants were 14 ± 3 (V1), 28 ± 3 (V2), 42 ± 4 (V3) and 91 ± 5 (V5) d old, and hospital visits took place when they were 56 ± 4 (V4), 119 ± 6 (V6), 182 ± 7 (V7) and 212 ± 7 (V8) d old. Mothers/carers kept records of the infants' diet, stool characteristics, symptoms of digestive tolerance and behaviour for 2 d before each visit. At each visit, the research team recorded anthropometric measurements and checked, and completed 2-d retrospective dietary, tolerance and health history questionnaires. Immunisation took place upon enrolment and approximately on days 56, 119 and 182. In brief, infants were immunised for hepatitis B at enrolment and on days 56, 119 and 182, diphtheria, tetanus and pertussis on days 56, 119 and 182 and Haemophilus influenzae type b on days 56 and 119, according to the NHMRC immunisation schedule(24). Blood was drawn for analysis of clinical biochemistry on day 119 to evaluate the effect of the test formulas before the introduction of solids. LC-PUFA status was also assessed at this time. Antibody response to immunisation was assessed on day 212, 28 days after the last immunisations for diphtheria, tetanus, pertussis and hepatitis B and 90 d after the last immunisation for H. influenzae type b.
Dietary intervention
Both formulas were whey predominant (70 % whey and 30 % casein) and contained protein, carbohydrates, fats, vitamins and minerals in quantities sufficient for the growth of healthy, term infants (Table 1). In addition, the experimental formula contained B. lactis (strain CNCM I-3446), fish oil DHA and AA (Arasoft®; Sofinol, Switzerland). The B. lactis strain was chosen because of its use in products for older infants and toddlers. The dose of DHA was based on the average composition in the breast milk of omnivorous women and to coincide with the dose used in other trials(Reference Marteau, de Vrese and Cellier15). We rationalised that the dose of AA should be equivalent to DHA to avoid an immune/inflammatory imbalance. Formulas were produced, blinded and supplied by the manufacturer (Nestlé, Konolfingen, Switzerland).
Table 1 Nutrient composition of the experimental and control formulas*
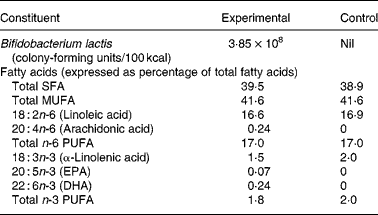
* Both formulas provided 1·83 g protein/100 kcal, 5·3 g fat/100 kcal and 11·2 g lactose/100 kcal.
Outcome measurements
The primary outcome was weight gain (g/d) from day 14 to day 119 to cover the period of exclusively feeding the test formulas. The secondary outcomes were recumbent length, head circumference, BMI, biochemical measurement of antibody titres, symptoms of digestive tolerance and occurrence of adverse events (AE). At each visit, anthropometric measurements were taken by one of three trained anthropometrists. Over 50 % of the assessments at day 14 and day 119 were done by a single person. Weight of infants was measured to the nearest 10 g on electronic scales, recumbent length was measured to the nearest 1 mm on standardised length boards and head circumference was measured to the nearest 1 mm using a standardised, non-elastic measuring tape. Evaluation of digestive tolerance was based on: volume of formula intake and any other dietary intakes; stool characteristics, including frequency (number of stools per 24 h), predominant colour (brown, yellow, green, red or black) and consistency (hard, formed, soft or liquid); frequency of spitting up or vomiting; behaviour (periods of restlessness or irritability that lasted for >30 min). AE were assessed at each visit by a nurse. AE were defined as illnesses or signs or symptoms occurring or worsening, or the occurrence of any abnormal laboratory findings during the course of the study and classified according to the International Classification of Diseases-10 criteria(25). AE were considered to be serious (SAE) if they were fatal or life-threatening events causing permanent harm or requiring or extending inpatient treatment at a hospital. The study investigators assessed the seriousness of an AE and causal relation to the study products.
Blood (2 ml) was drawn by venepuncture into heparinised tubes and centrifuged. Red cells were separated and washed three times with saline and lipids extracted for later fatty acid determination. The plasma was frozen at − 80°C. Determination of the concentration of Hb, albumin, prealbumin, total protein, urea, creatinine, ferritin, calcium, sodium, potassium, chloride, phosphate and glucose were performed at SouthPath Laboratory at Flinders Medical Centre. Titres of antibodies against diphtheria and tetanus were measured by ELISA and antibodies against hepatitis B were measured by Abbott Axsym (automated analyser) in the Department of Microbiology at the Women's and Children's Hospital. Antibodies against pertussis and H. influenzae type b were determined by ELISA at the National Centre for Immunisation Research and Surveillance, The Children's Hospital at Westmead(Reference Hawkes, Gibson and Roberton23). Lipids were extracted from red blood cells on the day of collection. Butylated hydroxyl anisole (0·005 % (w/v)) was added to the solvents (propanol and chloroform) used for lipid extraction. Total phospholipids were separated by TLC and the fatty acid profile was determined by capillary GC at the Child Nutrition Research Centre Laboratory at Flinders Medical Centre using previously described procedures(Reference Makrides, Hawkes and Neumann26).
Ethics
The present study was approved by the ethics committee of CYWHS and was conducted in accordance with the Declaration of Helsinki and its subsequent amendments. Written informed consent was obtained from the legal guardians of all infants. During the course of the study, we became aware that the genome of B. lactis (strain CNMI-3446) contained a gene that codes for tetracycline resistance. On report to the Research Ethics Committee, an independent expert review was commissioned. This review concluded that the B. lactis strain was safe based on the following:
the resistance was to an antibiotic that is not normally used in children;
tests to date have shown that the resistance gene does not spread to other bacteria in the gut;
the type of resistance is very common in a range of probiotic products available commercially;
the risks to the health of the child were negligible.
At the request of the Research Ethics Committee, a new patient information sheet was prepared containing this new information and participants already in the trial were re-consented. All but one continued to feed their assigned test formula.
Statistical methods
Sample size calculation was based on the primary outcome, which was weight gain. Growth of infants in the experimental and control formula groups was considered equivalent when the two-sided 90 % CI of the difference in the means of weight gain lay within the interval from − 3·9 to +3·9 g/d, a standard that has been used in previous trials(Reference Puccio, Cajozzo and Meli27). The sample size required to detect a difference in weight gain that is outside the − 3·9 to +3·9 g/d range based on an α-level of 0·05, a power (1 − β) of 0·9 and standard deviation of 6·1 g/d is forty-eight per group (power analysis and sample size calculations were performed by PASS 6.0; NCSS, Kaysville, UT, USA). This sample size is slightly lower than the Food and Drug Administration (FDA) requirement for clinical testing of infant formulas; therefore, we based the present study on the more conservative value of the FDA. According to the FDA guidelines, twenty-eight infants of each sex per group would have to be enrolled(28). Thus, fifty-six infants per group would have to complete the study. Allowing for a 20 % dropout, 140 infants had to be enrolled. The projected sample was also consistent with the number of infants involved in other dietary intervention trials assessing antibody titres to immunisations(Reference Hawkes, Gibson and Roberton23).
The intention-to-treat (ITT) analysis was performed on all randomised infants. The per protocol (PP) analysis excluded the data from infants who were withdrawn from the study, had a life-threatening event during the study, consumed >1 bottle per week of a different infant formula, did not consume the experimental formula for ≥ 7 consecutive days, were introduced to weaning food before 4 months of age, were hospitalised for ≥ 7 d or failed to complete the 14- or 119-d visit. The primary outcome (growth) was analysed in both the ITT and PP populations.
The mean difference in intake and growth measurements between the experimental and control groups was estimated by ANOVA correcting for sex. Growth measurements were also compared with the Centers for Disease Control and Prevention (CDC) references(Reference Kuczmarski, Ogden and Grummer-Strawn29). Blood analysis data were compared between groups using the t test (for normally distributed data) or Wilcoxon test (for skewed data). Stool counts for each infant were aggregated and divided by the number of days (counts/d), and the difference in frequency between the two groups was analysed by the t test. The number of days in which each stool colour and consistency occurred and the number of days in which infants were free of restlessness, flatulence, colic, spitting and vomiting were compared between groups using logistic regression. Finally, the incidence of AE (percentage of infants who had experienced at least one AE) was compared between groups with the exact χ2-test.
Statistical analysis was performed with SAS 8.02 (SAS Institute, Cary, NC, USA)
Results
Study population
One hundred and forty-two infants were enrolled and randomised to the experimental (n 72) or control group (n 70). There were no differences in the baseline characteristics between infants in the two study groups (Table 2). Ten (13·8 %) infants from the experimental group and eight (11·4 %) infants from the control group were withdrawn or lost to follow-up before the end of the study (Fig. 1). Twenty-seven infants did not follow the required dietary restriction (used non-study formulas or introduced solid food before 4 months of age) and therefore their data were excluded from the PP analysis. Since one infant in the experimental group dropped out only after the sixth visit (day 119 of age), the infant's data were available for growth analysis and the infant was kept in the PP population.
Table 2 Baseline characteristics of infants (intention-to-treat)*
(Mean values and standard deviations)
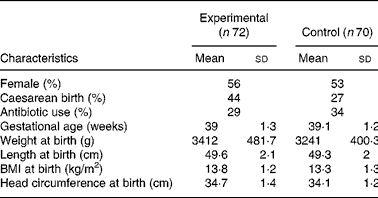
* There were no significant differences between groups.

Fig. 1 Trial profile. ITT, intention-to-treat; PP, per protocol.
Formula intake and growth
The mean volume of formula intake tended to be higher in the experimental group compared with the control group throughout the study (data not shown), but was significantly higher only on days 56 and 91 (mean and standard deviation for ITT: 853 (sd 169) v. 785 (sd 175) ml/d, P = 0·04 and 939 (sd 169) v. 855 (sd 183) ml/d, P = 0·01, respectively; for PP: 855 (sd 162) v. 771 (sd 168) ml/d, P = 0·02 and 936 (sd 172) v. 866 (sd 185) ml/d, P = 0·05, respectively). The mean daily volume of formula intake over the course of the entire study was also significantly higher in the experimental group compared with the control group (ITT: 873 (sd 152) ml/d v. 799 (sd 151) ml/d, t test P = 0·001; PP: 893 (sd 130) v. 830 (sd 134), P = 0·014).
There were no significant differences in weight between the two groups throughout the study (data not shown). Mean weight gain (from day 14 to day 119) between the experimental and control groups was 1·5 and 2·0 g/d for the ITT and PP populations, respectively. The 90 % CI (ITT: − 0·08–3·1 g/d and PP: 0·1–3·8 g/d in the PP population) lay within the − 3·9 to +3·9 g/d interval, indicating equivalent growth among infants in the two groups. Similarly, the mean changes in length, head circumference and BMI were not significantly different between the two groups (Table 3). A comparison of weight-for-age, length-for-age and head circumference-for-age with the CDC growth references showed that z-scores were within the normal ranges for both groups (data not shown).
Table 3 Mean changes in anthropometric measurements during the study (per protocol)*
(Mean values and standard deviations)
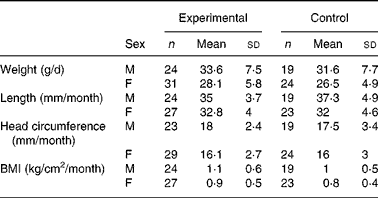
M, males; F, females.
* Treatment differences of anthropomorphic measurements were estimated by a mixed model corrected for sex. There were no significant differences between groups.
Biochemical and immunological analyses
On day 119, the concentrations of DHA, AA and EPA in red blood cell membranes were significantly higher among infants in the experimental group compared with those in the control group (Table 4). On the other hand, the concentrations of linoleic acid and α-linolenic acid were higher in infants in the control group. Both results are reflective of the fat composition of the formulas (Table 1).
Table 4 Fatty acid concentration (percentage of total phospholipid fatty acid) in erythrocyte membranes on day 119 (per protocol population)
(Mean values and standard deviations)
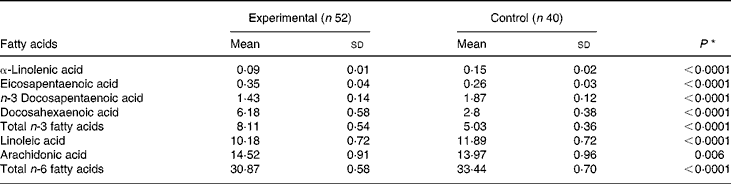
Comparison by the t test; P < 0·05 was considered significant.
None of the standard blood biochemical measurements differed between the two groups (data not shown), except that the measurements of blood glucose and albumin concentrations were higher in the control group (4·7 (sd 0·5) v. 4·4 (sd 0·7) mmol/l, P = 0·012 for glucose and 40·8 (sd 2·5) v. 39·5 (sd 2·9) g/l, P = 0·03 for albumin). However, all values are within the normal range for infants.
On day 212, there were no differences between the control and experimental groups in the geometric mean titres of antibodies against diphtheria (0·82 v. 0·92 IU/ml, respectively), H. influenzae type b (2·23 v. 2·01 mg/ml, respectively), hepatitis B (27·11 v. 24·53 IU/ml, respectively), pertussis filamentous haemagglutinin (3·67 IU/ml for both groups), pertussis pertactin (4·06 IU/ml for both groups), pertussis toxin (2·46 v. 2·23 IU/ml, respectively) and tetanus (1·11 IU/ml for both groups).
Digestive tolerance
In general, stool characteristics were similar between the two groups (data not shown). However, green stools occurred significantly less frequently (25·6 (sd 22·5) v. 33·3 ± 25·5 %, P = 0·002) and yellow stools occurred significantly more frequently (67·6 (sd 25·2) v. 50·3 ± 27·3 %, P < 0·001) among infants in the experimental group. Stools, colic, spitting up, vomiting and restlessness occurred at similar frequencies in the two groups (data not shown).
Adverse events
During the study, 403 AE were reported in 124 infants (sixty in the experimental and sixty-four in the control groups) in the ITT population (Table 5). The most common AE were infections, dermatitis, digestive problems and feeding problems (vomiting during or right after feeding), and only the frequency of feeding problems was significantly lower in the experimental group compared with the control group (P = 0·03). Forty AE in twenty-nine infants (eighteen in the experimental and eleven in the control groups) were reported as serious (SAE), and were similarly distributed among the two groups (P = 0·21). All SAE required hospitalisation of infants. The most frequently occurring SAE were respiratory problems (seven in the experimental and four in the control groups) and gastrointestinal problems (three in each group). Most of the SAE were considered to be unrelated (n 20) or unlikely to be related (n 17) to the formulas. Three SAE considered to be probably related to the formulas were gastrointestinal problems (one in each group) and a respiratory problem in the control group. There were no deaths.
Table 5 Adverse events (percentage of affected infants) occurring in the intention-to-treat population and classified according to the International Classification of Diseases-10 criteria

AE, adverse events.
* No significant difference among groups by the χ2-test except P < 0·05.
Discussion
In the present study, we evaluated the safety of a formula containing the probiotic B. lactis and the LC-PUFA DHA and AA, by comparing the weight gain of infants fed the experimental formula with that of infants fed a similar formula lacking these components. There was no difference in weight gain between infants in the two groups during the study period (day 14–day 119). Other growth parameters also showed no differences at any of the time points, confirming that growth was similar in the two groups. While individual components such as B. lactis or LC-PUFA are added for benefit, it is important that the combination of such novel components is also tested for safety.
It has previously been reported that decreases in the concentration of plasma AA in preterm infants, due partly to the addition of n-3 LC-PUFA in infant formulas, correlated with a decrease in growth(Reference Carlson, Werkman and Peeples30). Subsequently, it has been proposed that n-3 LC-PUFA supplementation should occur in conjunction with AA to avoid lowering the plasma and erythrocyte concentrations of AA. This has led to a discussion relating to the best ratio of n-3 LC-PUFA and AA for infant formula, and recommendations suggesting that a 1:2 ratio may be optimal. Our earlier trial assessing DHA and AA at a concentration of 0·34 % of total fatty acids in a 1:1 ratio(Reference Makrides, Neumann and Simmer31) as well as the present study assessing a concentration of 0·23 % of DHA and AA in a 1:1 ratio both demonstrate equivalent growth and increase the erythrocyte concentrations of both DHA and AA relative to the control, unsupplemented infant formula. These data imply that a 1:1 ratio of DHA to AA does not result in a reduction in AA status across different concentrations. A 1:1 ratio of DHA to AA also falls within the range of human milk values reported worldwide and summarised in a recent review(Reference Brenna, Varamini and Jensen32). Furthermore, recent analyses of randomised trials with term infants receiving formula supplemented with only n-3 LC-PUFA (and causing a reduction in infant AA status) have not shown any effect on growth(Reference Makrides, Gibson and Udell5, Reference Simmer and Patole21). Addition of probiotics to infant formulas has not been associated with any reports of growth effects in term infants. However, the effect of combining LC-PUFA and probiotics was not known, although there is no reason to believe a priori that delivery of the combination of these two components would affect growth.
Das(Reference Das22) proposed that the combination of LC-PUFA and probiotics may be beneficial, since LC-PUFA help to colonise the nascent microbiota of infants by probiotics and enhance adherence of probiotics to gut mucosal cells. LC-PUFA themselves have been shown to modulate components of the immune system. Field et al. (Reference Field, Thomson and Van Aerde7) showed that dietary fatty acids have an effect on several functional indices of immune development (increased concentration of antigen-mature T-cells and increased production of IL10) in preterm infants. Other possible effects include stimulating lymphocyte proliferation, cytokine production, natural killer cell activity and antigen presentation (reviewed in Field et al. (Reference Field, Clandinin and Van Aerde33)).
A comparison of titres of IgG against diphtheria, H. influenzae type b, hepatitis B, pertussis and tetanus vaccines when infants were approximately 7 months of age (1 month after they had completed a primary immunisation course) showed that there were no differences between the two groups. The present study was powered to detect differences in weight gain and may not have been sufficiently powered to detect differences in IgG concentrations following immunisation, although other trials of formula feeding have shown differences in vaccine titres with similar numbers of infants(Reference Hawkes, Gibson and Roberton23). Since infants mount a strong response to vaccines, especially after several doses, it may be difficult to observe subtle effects of LC-PUFA and probiotics on the immune response. Measurement of antibody titres after the first immunisation may give a better indication of the effect on the immune response. Alternatively, effects on the immune response may be more evident for vaccines that are delivered orally or via other mucosal routes. Indeed, de Vrese et al. (Reference de Vrese, Rautenberg and Laue34) showed that probiotics enhanced the antibody response to orally delivered polio vaccine. A different group studying the effect of probiotics on the vaccination response in infants suggested that probiotics may improve the immune response to H. influenzae type b vaccine, whereas there was no indication of any effect on the antibody response to diphtheria and tetanus vaccines(Reference Kukkonen, Nieminen and Poussa35).
Interestingly, fewer parents with infants in the LC-PUFA and probiotic group reported feeding problems of the newborn compared with the control. This International Classification of Diseases-10 classification mostly describes vomiting during or directly after a feed that is not associated with fever or other gastrointestinal symptoms such as diarrhoea. Whether this finding indicates improved tolerance in infants fed the experimental formula is not clear because our sample size was too small to exclude the possibility of random error.
In the present study, over 50 % of families with eligible infants consented to participate and, of these, 87 % completed the trial. The high participation rate and retention rate for this type of trial point towards a robust internal and external validity of the trial data, which demonstrate that an experimental formula with LC-PUFA and probiotics promotes normal growth of term infants. Additional studies are required to determine whether the combination of LC-PUFA and probiotics has any functional effect on the infants' immune system.
Acknowledgements
Nestlé Nutrition provided the formulas and funding to conduct the trial. Salary support for M. M. and R. A. G. was from Senior Research Fellowships of the NHMRC of Australia. We thank Heather Garreffa, Jo Collins, Karen Bellchambers, Dr Andy McPhee, Diana Weber, Michelle Clark and Dr Jan Walker for their clinical, administrative and technical support, Dr Tamara Zutlevics and Prof. John Turnidge for their ethical review. We also thank Dr Andrew Lawrence and Mark Hanlon for serological analysis. We also acknowledge the support of Dr Henri Dirren and Dr Philippe Steenhout, and Choo Hoffmann who undertook independent source data verification and Dr Makda Fisseha for providing medical writing services on behalf of HPM Geneva SA. All authors participated in the design of the trial. D. B. and M. M. prepared the data collection materials, and data collection was under the supervision of M. M. and R. A. G. H. M. supervised the vaccine analyses. J. M. performed the data analysis. All authors were involved in the interpretation of the data, commented on drafts of the manuscript and approved the final version. D. B., J. M. and J. C. M. are employees of Nestlé. R. A. G., M. M. and H. M. declare that there is no conflict of interest.






