


N-3 long-chain (≥C20) PUFA (LC-PUFA) are fatty acids that contain more than two double bonds, with the first located three carbons from the methyl end group(Reference Tocher and Glencross1). The most common LC-PUFA are EPA (20 : 5n-3, EPA) and DHA (22 : 6n-3, DHA). The n-3 LC-PUFA reportedly play numerous positive roles in a range of human pathologies, including obesity, CVD and cancer, and have beneficial effects on nervous system development(Reference Tocher2). Dietary intake of n-3 LC-PUFA is necessary for humans to satisfy certain physiological demands due to a limited ability to convert α-linolenic acid to n-3 LC-PUFA(Reference Bernna3). Fish, especially marine fish, serve as a main source of n-3 LC-PUFA for humans(Reference Tur, Bibiloni and Sureda4). The n-3 LC-PUFA are also essential fatty acids for fish (particularly marine fish)(Reference Tocher5), and presently, fishmeal and fish oil are almost indispensable as components of farmed marine fish feeds to compensate for the deficiency in n-3 LC-PUFA synthesis by the fish themselves(Reference Sargent, Tocher and Bell6). The reliance on finite marine resources derived from capture fisheries is unsustainable; therefore, the development of methods to improve the n-3 LC-PUFA content in farmed fish is urgently required for both aquaculture and human consumers.
An effective way to increase the n-3 LC-PUFA content in fish is to enhance the uptake and deposition of dietary n-3 LC-PUFA. In fish, the processes of uptake and deposition of dietary fatty acids resemble those in mammals, that is mainly including the emulsification of fat and catabolism of emulsified fat(Reference Kjær, Vegusdal and Berge7). Among them, fatty acid-binding proteins (FABP), which are involved in the intracellular transport of fatty acids, have been reported to be directly related to the fatty acids content. In human (Homo sapiens) Caco-2 cells, the knockdown of the fabp1 gene resulted in a decrease in the uptake and accumulation of oleic acid(Reference Luciana, Ban and Natalia8). Analogously, the gene expression of fabp5 affects the DHA and EPA levels in rat (Rattus norvegicus) PC12 cells and H. sapiens hCMEC/D3(Reference Liu, Almague and Bu9–Reference Pan, Short and Choy11). In vivo, experimental mice (Mus musculus) showed decreased linoleic acid uptake and accumulation after the fabp3 gene was knockout compared with the wild-type M. musculus (Reference Islam, Kagawa and Sharifi12). In fish, the gene expression of fabps has been shown to be associated with dietary fatty acids. For example, the fabp3 mRNA level in the myosepta of Atlantic salmon (Salmo salar) fed vegetable oil was significantly lower compared with those fed fish oil(Reference Torstensen, Nanton and Olsvik13). In addition, the hepatic fabp1 gene expression was considerably increased in Japanese seabass (Lateolabrax japonicus) fed arachidonic acid(Reference Xu, Zhang and Wang14). These studies suggest the potential feasibility of improving the n-3 LC-PUFA content of farmed fish by regulating the gene expression of fabps.
Arguably, the uptake and deposition of n-3 LC-PUFA in the muscle, that is the edible portion, are the focus in aquaculture nutritional studies, with the potential to improve the quality of meat. The liver, another important tissue is organisms, is widely involved in fatty acid synthesis(Reference Kolditz, Paboeuf and Borthaire15) and is, therefore, rarely associated with fatty acid deposition. Indeed, studies on grass carp (Ctenopharyngodon idella)(Reference Ji, Li and Liu16) and gibel carp (Carassius auratus gibelio)(Reference Chen, Zhu and Han17) have suggested that the liver has certain abilities to deposit fatty acid (mainly n-3 LC-PUFA) without having negative effects on growth performance and antioxidant markers. Besides, being the ‘transit hub’ of substance metabolism(Reference Tocher and Glencross1), the fatty acid levels of the liver and the whole body are inseparable. Therefore, liver n-3 LC-PUFA uptake and deposition are, arguably, very noteworthy, despite the paucity of studies of these processes.
Golden pompano (Trachinotus ovatus) is an important concerned marine fish by the Ministry of Agriculture of China for mariculture with a considerable market foreground. T. ovatus have been demonstrated to have little or no ability to biosynthesise n-3 LC-PUFA in our recent study(Reference Zhang, Chen and You18). Previously, we found that T. ovatus hepatic n-3 LC-PUFA content increased with increasing dietary n-3 LC-PUFA content within a certain range. Meanwhile, transcriptome analysis of the liver indicated that the fabp4 gene expression was positively correlated with the hepatic n-3 LC-PUFA content; therefore, fabp4 may be a candidate gene for regulating the n-3 LC-PUFA content(Reference Lei, Li and Tian19). Based on these findings, we aimed to validate and characterise the potential regulatory role and mechanism of T. ovatus fabp4 on the n-3 LC-PUFA content. Our findings provide reference data for the reduction of fish oil and fishmeal supplement levels in marine fish feed.
Materials and methods
Animal study ethics
All procedures performed on fish were in accordance with the National Institutes of Health guide for the care and use of laboratory animals (NIH Publications No. 8023, revised 1978) and approved by the Institutional Animal Care and Use Committee of South China Agricultural University (Guangdong, China).
Trachinotus ovatus genomic DNA extraction and cloning of the 5′ flanking sequence of fatty acid-binding protein 4
A live T. ovatus with unknown sex weighing approximately 500 g was randomly purchased from a local commercial market for the experiment. The fish was raised without food in a 320-litre aquarium for 4 h under natural conditions (dissolved oxygen was provided). Before sampling, the anaesthetic (2-phenoxyethanol; Sigma-Aldrich) was mixed with water in a 50-ml volumetric flask and then further diluted with water to a concentration of 0·01 % (w/v) in a bucket (17 litres) for the anaesthesia of the test fish. When the fish showed no response to external touch or stimuli and total loss of equilibrium, it was immediately removed from the bucket and the liver tissue was harvested. Genomic DNA was extracted from the liver tissue using a commercial kit (Beyotime Biotechnology Co. Ltd) according to the manufacturer’s recommendation. The obtained DNA sample was electrophoresed on a 1 % (w/v) agarose gels to check its integrity, and the DNA was quantified using a spectrophotometer (NanoDrop 1000; Thermo Scientific). The fabp4 gene sequence was referenced from our nonpublished whole-genome sequencing data of T. ovatus. Specific PCR primers (online Supplementary Table S1) were then designed to amplify the fabp4 gene 5′ flanking sequence.
The PCR amplification mixture consisted of DNA, upstream primer, downstream primer, 2 × Taq PCR Mix (TianGen Biotech) and sterilised double-distilled water at 2 μl (50 ng), 2 μl, 2 μl, 25 μl and 19 μl, respectively. The amplification procedure comprised three steps: initial denaturation at 94°C for 3 min; 35 cycles of 94°C for 30 s, 62°C for 30 s and 72°C for 1 min; and final extension at 72°C for 5 min. The PCR products were then electrophoresed in a 1 % (w/v) agarose gel, and the bands of expected sizes were purified using a commercial kit (TianGen Biotech). The obtained fragments were then cloned into a pMDTM19-T (TaKaRa) vector following the manufacturer’s instructions. The ligation mixtures were transformed into Escherichia coli DH5α competent cells (TianGen Biotech). A plasmid preparation kit (TianGen Biotech) was used to purify the plasmids containing inserts of an expected size. The obtained fragment was then sequenced at Sangon Biotech.
Cell culture
The HEK 293T cell line was purchased from the Chinese Type Culture Collection (Shanghai, China). The medium for cell growth composed of Dulbecco’s modified Eagle’s medium/nutrient mixture F12 (Gibco), 10 % fetal bovine serum (Gibco), penicillin (100 U/ml; Beyotime Biotechnology Co. Ltd) and streptomycin (100 U/ml; Beyotime Biotechnology Co. Ltd). The cells were incubated at 37°C with 5 % CO2, and the medium was changed every 2–3 d.
The Epinephelus coioides hepatocyte line was obtained from our laboratory, where it was successfully established without publication. The Epinephelus coioides hepatocyte line growth medium shared the same composition as that of HEK 293T cells. Differently, the fetal bovine serum used in Epinephelus coioides hepatocyte line culture was obtained from Clark (Clark Bioscience Co. Ltd). The cells were maintained at 28°C without CO2.
RNA isolation and quantitative real-time PCR
Total RNA was extracted from the cells using RNAiso Plus (TaKaRa) following the manufacturer’s recommendation. Prior to this, the cells were collected using 0·25 % trypsin and centrifuged at 1500 rpm for 3 min to form pellets in 1·5 ml centrifuge tubes. The integrity of RNA samples was determined using 1 % (w/v) agarose gel electrophoresis, and the quantification of RNA samples was detected using a spectrophotometer (NanoDrop 1000; Thermo Scientific). Prior to quantitative real-time PCR detection, 1 μg of total RNA was reverse-transcribed using a PrimeScript® RT reagent kit (TaKaRa). The gene transcript level was measured in triplicate (CFX 96 Real-Time PCR Detection System; Bio-Rad), and the amplification mixture contained 0·8 μl of each primer (10 μm) (online Supplementary Table S1), 1 μl (25 ng) of first-strand cDNA product, 10 μl of 2 × SYBR® Premix Ex TaqTMII (TaKaRa) and 7·4 μl sterilised double-distilled water. The amplification conditions began with 30 s at 95°C, followed by 40 cycles of 5 s at 95°C, 45 s at 62°C and 1 min at 72°C. The melting curve was used as a criterion to determine that a single product was amplified. The relative gene expression was normalised with that of the reference gene (β-actin). The comparative CT method (2−ΔΔCt)(Reference Livak and Schmittgen20) was introduced to calculate the results that expressed relative to the control group. In the quantitative real-time PCR of fabp4 and β-actin, the PCR product sequences shared a close identity with that of E. coioides. Consequently, the basic expression levels of fabp4 and β-actin were detected. In contrast, the sequence of the PPAR-γ quantitative real-time PCR product shared a low identity with that of E. coioides and the basic expression level of PPARγ was not detected. For calculation purposes, we artificially set the expression level of the control group to 1.
Fatty acid composition analysis
The cellular lipids were extracted, and the fatty acid profile was assessed according to our previous methods(Reference Li, Hu and Zheng21,Reference Li, Monroig and Zhang22) . The cells were digested and collected using 0·25 % trypsin after washing twice with PBS (pH = 7·3 (sd 0·1), Beyotime Biotechnology Co. Ltd). The cells were then mixed with chloroform/methanol (2:1, by vol) containing 0·01 % butylated hydroxytoluene with gentle shaking to extract the total lipids. Distilled water was added, then the mixture was centrifuged at 3000 rpm for 5 min and the supernatant was discarded. Subsequently, potassium hydroxide methanol (0·5 m) was used to saponify samples at 65°C, and hexane was then used to dissolve the samples. Finally, the fatty acid methyl esters in the upper layer were separated using centrifugation and analysed using GC (Agilent 7890B; Agilent Technologies). Individual methyl esters were identified by comparison with authentic standards (Sigma-Aldrich). The results were expressed as the percentages of each fatty acid in the total fatty acids.
Plasmid constructions and transient transfection
The complete open reading frame of T. ovatus fabp4 was successfully cloned in our previous study(Reference Lei, Li and Zhang23) and amplified using 2 × Taq PCR Mix (TianGen Biotech) as described above. The pcDNA3.1 (+) vector (Invitrogen) was used to construct the recombinant plasmid which carries the fabp4 gene (pcDNA3.1-fabp4). Another overexpression recombinant plasmid, pcDNA3.1-PPARγ was provided by Dr Meng Li of our research team. The accuracy of recombinant plasmid was verified using sequencing (Sangon Biotech Co. Ltd). The primers used are shown in online Supplementary Table S1.
A commercial endofree maxi plasmid kit (TianGen Biotech) was used to purify the pcDNA3.1-fabp4 and pcDNA3.1-PPARγ recombinant plasmids, and the X-tremeGene HP DNA transfection reagent (Roche) was used for cell transfection after the cells reached 70 % confluence. The culture medium was replaced with fresh medium without antibiotics prior to cell transfection. The ratio of X-tremeGene HP DNA transfection reagent (μl):DNA (μg) was 2:1.
According to the sequence from the fabp4 gene 5′ flanking sequence cloning, corresponding primers (online Supplementary Table S1) were designed to obtain the accurate sequence of the full-length promoter fragment (D1) and another five progressive deletion fragments (D2–D6). These six fragments were amplified using 2 × Taq PCR Mix (TianGen Biotech) with genomic DNA as the template with PCR amplification conditions as described above. pGL4.10 (Promega) and pGL4.75 (Promega) were used as carrier plasmid and reference plasmid, respectively. Details regarding the construction of the recombinant plasmid were mentioned in our previous studies(Reference Dong, Wang and Chen24,Reference Dong, Zhao and Chen25) . The commercial endofree maxi plasmid kit (TianGen Biotech) was used to extract the recombinant plasmids for further transfection. Previously, these recombinant plasmids were also sequenced (Sangon Biotech Co. Ltd) to ensure accuracy.
Site-directed mutagenesis of the potential regulator of fatty acid-binding protein 4
To further investigate the potential regulator of fabp4, site-directed mutagenesis was conducted based on overlap extension PCR. The site-directed mutant was designed from the deletion mutant D6 (pGL4.10-D6), which targets the transcription factor PPARγ. Two pairs of primers (F1 and R1, F2 and R2) were designed (online Supplementary Table S1). Primers F1 and R1 occur at either ends of the sequence, whereas F2 and R2 are positioned internally. The mutation points were introduced in the F2 and R2 primer pair. First, pGL 4.10-D6 was used as the template for PCR amplification in accordance with the patterns of ‘F1 + R2’ and ‘F2 + R1’ in a 50 μl amplification volume, containing 2 μl of each primer (10 μm), 50 ng (2 μl) of template, 25 μl of 2 × Pfu PCR Mix (TianGen Biotech) and 19 μl sterilised double-distilled water. After purification with a commercial kit (TianGen Biotech), the products were mixed at equal volumes. Subsequently, the mixed product was used as the template, and F1 and R1 were used as primers for amplification in a 50 μl volume, containing 2 μl of each primer (10 μm), 2 μl (50 ng) of template, 25 μl of 2 × Taq PCR Mix (TianGen Biotech) and 19 μl of sterilised double-distilled water. The Mastercycler® nexus gradient PCR system (Eppendorf) was programmed for touchdown PCR under the following conditions: 94°C for 3 min, 94°C for 30 s, 72°C for 30 s followed by 30 cycles of 1-min step: the initial annealing temperature of 72°C was decreased by 0·5°C per cycle to reach 57°C for the final cycle. The mixture was then subjected to 94°C for 30 s, 57°C for 30 s and 72°C for 1 min for further fifteen cycles. The PCR ended with an extra extension of 5 min at 72°C. After the amplification, electrophoresis of 10 μl samples in 1 % (w/v) agarose gels detected the unique PCR product. This was excised, isolated (TianGen Biotech), inserted into pMDTM19-T (TaKaRa) and sequenced at Sangon Biotech. Purification of the site-directed mutant was performed using the above-mentioned endofree maxi plasmid kit (TianGen Biotech).
Dual-luciferase reporter assay
The dual-luciferase assay was performed after the HEK 293T cells reached 70 % confluence to determine the activity of a given promoter. The reporter firefly luciferase construct and reference plasmid pGL4.75 (Promega) were co-transfected into the cells at a ratio of 10 000:1 (w/w) using Lipofectamine® 2000 Reagent (Invitrogen) according to manufacturer’s recommendation. Furthermore, empty pGL4.10 plasmids were used as the negative control. The plasmids were incubated with cells for 24 h; then, a fresh culture medium without plasmids was added to replace the original medium. After another 24 h (i.e. 48 h after transfection), luciferase assays were performed using a dual-luciferase reporter assay system (Promega). The results were expressed as the luminescence ratio of firefly:renilla luciferase. Twelve wells were included in one treatment group, and three dual-luciferase reporter assays were conducted independently.
Statistical analysis
All data are expressed as mean and standard deviations. The data from the core promoter region identification and luciferase assays after transfection with different concentrations of pcDNA3.1-PPARγ were verified using a one-way ANOVA followed by Duncan’s post hoc test using PASW Statistics version 18 (SPSS). All other data were tested by comparing groups using an independent-samples t test in PASW Statistics version 18 (SPSS). Tests for normality and homoscedasticity were performed prior to the formal statistical analysis.
Results
Cloning of the 5′ flanking sequence of fatty acid-binding protein 4 in Trachinotus ovatus and activity analysis
In this study, 2059 bp (from the initiation codon ATG) of the 5′ flanking sequence of the fabp4 gene in T. ovatus was cloned and treated as the promoter candidate (Fig. 1). The first base of the initiation codon ATG was defined as +1 in the sequence. Five progressive deletion fragments were constructed, namely D1 (−2 bp to −2006 bp), D2 (−2 bp to −1521 bp), D3 (−2 bp to −1158 bp), D4 (−2 bp to −733 bp) and D5 (−2 bp to −241 bp). The dual-luciferase reporter analysis indicated that deletion of the fragment from −2006 bp to −1521 bp caused a decrease in promoter activity compared with that of the original promoter candidate (D1). No significant differences were found in promoter activity after the sequences between −1521 bp to −1158 bp and −1158 bp to −733 bp were deleted. However, the deletion of fragment from −733 bp to −241 bp resulted in an increase in the promoter activity. These results indicated that the core promoter region was located between −2006 bp and −1521 bp (Fig. 2).

Fig. 1. The 5′ flanking sequence of the Trachinotus ovatus fatty acid-binding protein 4 (fabp4) gene and partial predicted binding sites of transcription factors (red: Forkhead box O1 (FoxO1); orange: CCAAT/enhancer binding protein (C/EBPα); green: PPARα; blue: PPARγ). Bold-faced bases indicate the initiation codon.
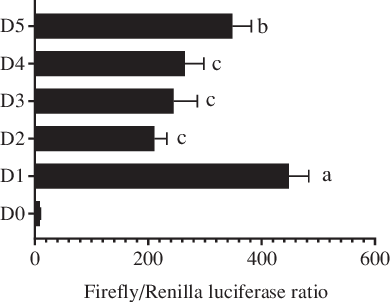
Fig. 2. Analysis of Trachinotus ovatus fatty acid-binding protein 4 (fabp4) gene promoter activity. D1–D5 indicates the five progressive deletion fragments, namely −2 bp to −2006 bp, −2 bp to −1521 bp, −2 bp to −1158 bp, −2 bp to −733 bp and −2 bp to −241 bp (The first base of the initiation codon ATG was defined as +1). D0 represents the negative control without the promoter region (pGL4.10-empty). Values are expressed as means and standard deviations. Means at a time without a common symbol are significantly different (P ≤ 0·05).
Fatty acid-binding protein 4 promoted the uptake of DHA in hepatocytes
In our previous study on T. ovatus, we found that the hepatic mRNA level of fabp4 increased with increasing n-3 LC-PUFA content in the diet and in the experimental fish liver (online Supplementary Fig. S1) and fabp4 was identified as a candidate gene for the regulation of the n-3 LC-PUFA content(Reference Lei, Li and Tian19). Herein, we further verified the role of fabp4 in regulating the n-3 LC-PUFA content in vitro. Compared with the control group, the DHA content increased significantly in the fabp4 overexpression group and the content of DHA was decreased after the fabp4 gene was suppressed by BMS309403 (inhibitor of fabp4; MedChemExpress) (Fig. 3). The changes in the other fatty acids are shown in online Supplementary Fig. S2.
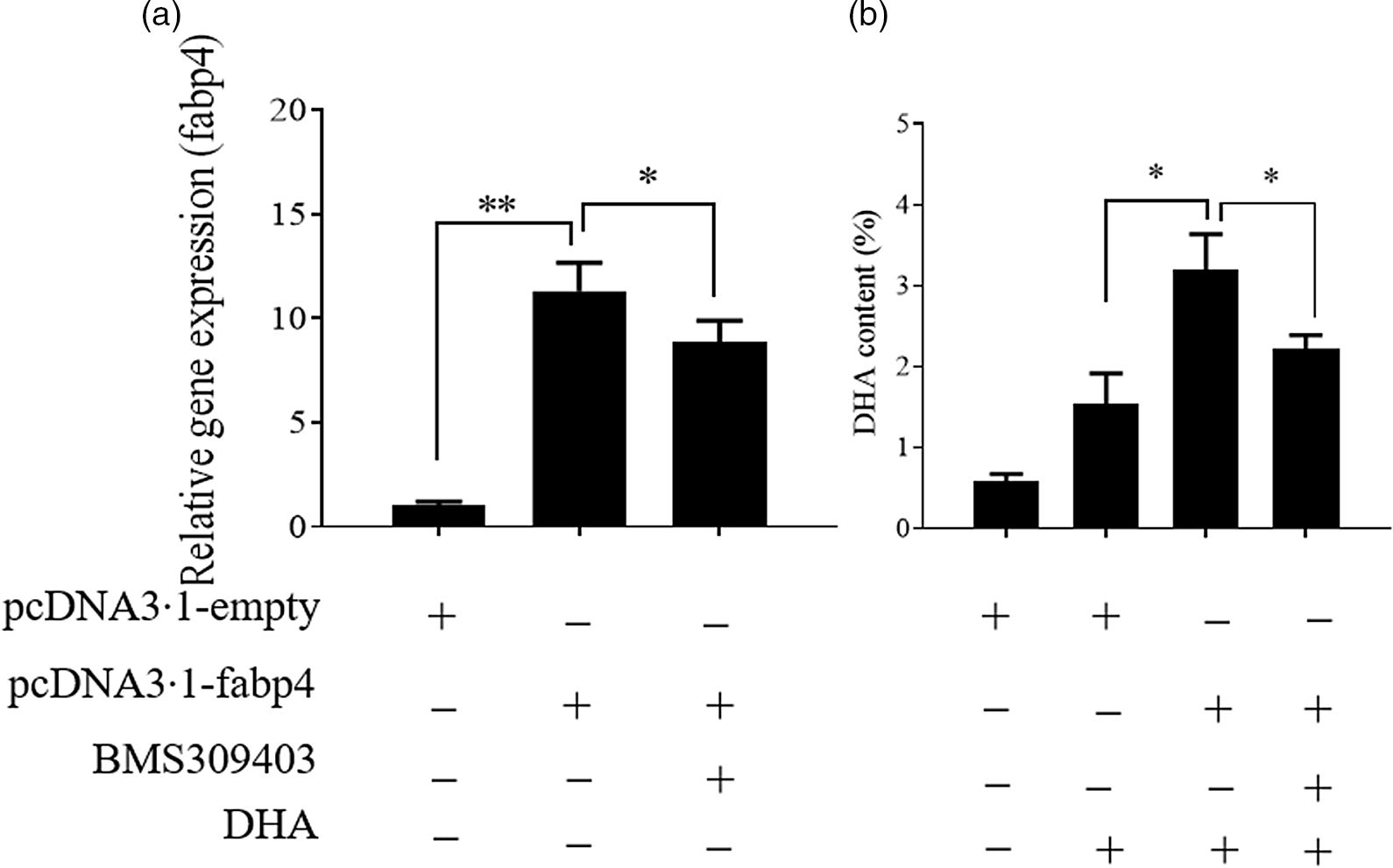
Fig. 3. Fatty acid-binding protein 4 (Fabp4) promotes the uptake of DHA. Values are expressed as means and standard deviations (n 3). The Epinephelus coioides hepatocyte line (ECHL) cells were transfected with pcDNA3.1-fabp4 or pcDNA3.1-empty plasmids for 12 h. Subsequently, 50 μm DHA and 75 μm BMS309403 were added as mentioned for another 24 h (BMS309403 was added 2 h prior to DHA) without recombinant plasmids. Cells were harvested for the detection of fabp4 gene expression (a) DHA content (b). *, P ≤ 0·05; **, P ≤ 0·01.
Stimulation of PPARγ contributed to the uptake of DHA in hepatocytes
Previously, we found that the change in PPARγ gene expression was consistent with the change in hepatic n-3 LC-PUFA content (not published) in T. ovatus fed with n-3 LC-PUFA (0·64–2·30 %). Meanwhile, transcriptome analysis of T. ovatus liver also showed that PPARγ was a potential regulator of the n-3 LC-PUFA content(Reference Lei, Li and Tian19). In this study, the uptake of DHA was markedly enhanced after the PPARγ gene was up-regulated by overexpression. Conversely, an antagonist of PPARγ, GW9662 (Sigma-Aldrich) attenuated an increase in the DHA content by inducing PPARγ overexpression (Fig. 4). The changes in the other fatty acids are shown in online Supplementary Fig. S2.
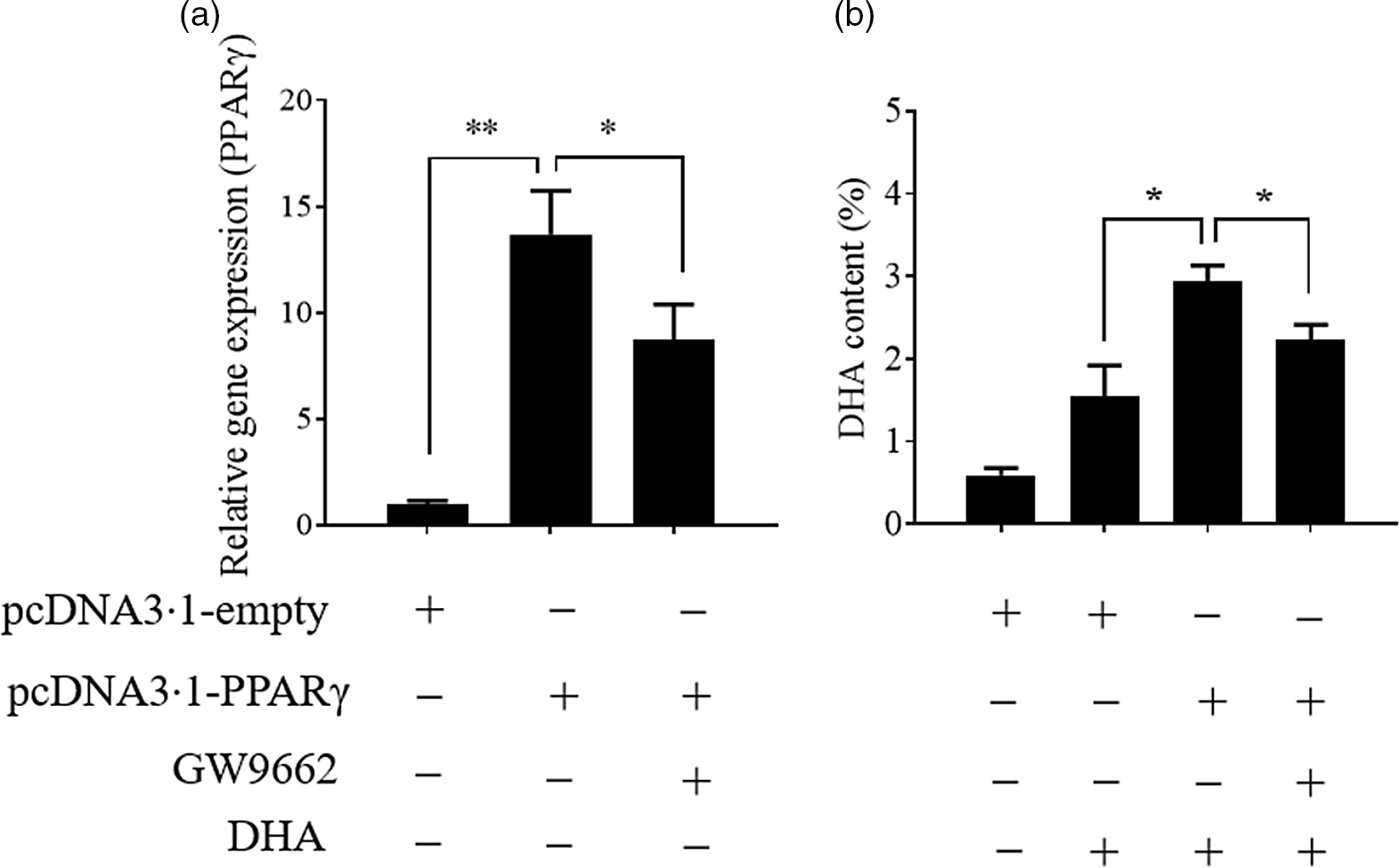
Fig. 4. Stimulation of PPARγ contributes to the uptake of DHA. Values are expressed as means and standard deviations (n 3). The Epinephelus coioides hepatocyte line (ECHL) cells were transfected with pcDNA3.1-empty or pcDNA3.1-PPARγ plasmids. After 12 h, the plasmids were removed and the cells were incubated with 50 μm DHA and 75 μm GW9662 as shown (GW9662 was added 2 h prior to DHA) for another 24 h. Cells were collected to measure PPARγ gene expression (a) and DHA content (b). *, P ≤ 0·05; **, P ≤ 0·01.
PPARγ regulated the expression of fatty acid-binding protein 4 at the transcriptional level
To identify possible transcription factor binding sites in the core promoter region of the T. ovatus fabp4 gene, we used online software to search for the potential binding site (http://bioinfo.life.hust.edu.cn/hTFtarget#!/prediction). Many transcription factors, such as Forkhead box O1, CCAAT/enhancer binding protein and transcription factor 5 binding sites, were identified in the core promoter region of T. ovatus fabp4 gene with high scores (Fig. 1). According to our previous study, PPARγ is a potential regulator of the T. ovatus fabp4 gene and two PPARγ binding sites were predicted in the present study (online Supplementary Fig. S3). To determine whether PPARγ regulates fabp4 gene expression, a site-directed mutant was constructed and transfected into HEK 293T cells for detection of luciferase activity. As shown in Fig. 5(a), the mutation of the PPARγ binding site (with the highest score) resulted in a significant decrease in luciferase activity. To further confirm the role of PPARγ in fabp4 expression, HEK 293T cells were co-transfected with D1, together with different concentrations of pcDNA3.1-PPARγ. The results showed that pcDNA3.1-PPARγ increased the luciferase activity in a dose-dependent manner compared with the control group (Fig. 5(b)). These findings strongly suggest that T. ovatus fabp4 may be a target of PPARγ.

Fig. 5. Fatty acid-binding protein 4 (Fabp4) in Trachinotus ovatus is a target of PPARγ. Values are expressed as means and standard deviations. (a): Luciferase activity was detected in HEK 293T cells that were transfected with pGL4.10-empty (D0), wild-type D6 (wild D6) or mutant D6 for 24 h. *, P ≤ 0·05. (b): HEK 293T cells were co-transfected with the fabp4 full-length promoter fragment (D1) and different concentrations of pcDNA3.1-PPARγ for 24 h. Means at a time point without a common symbol are significantly different (P ≤ 0·05).
PPARγ regulated the fatty acid-binding protein 4-mediated uptake of DHA in hepatocytes
As shown in Fig. 6, the simultaneous overexpression of PPARγ and fabp4 increased the mRNA levels of PPARγ and fabp4 significantly, by about 15-fold and 16-fold, respectively, compared with the empty plasmid group. An antagonist of fabp4, BMS309403 effectively reduced the expression of fabp4 with no significant effect on the mRNA level of PPARγ (Fig. 6(a)). Meanwhile, the simultaneous overexpression of PPARγ and fabp4 markedly enhanced the DHA content compared with the empty plasmid group. However, suppression of fabp4 via BMS309403 attenuated the increase in the DHA content (Fig. 6(b)). Conversely, treating Epinephelus coioides hepatocyte line cells with GW9662 markedly attenuated the up-regulation of PPARγ induced by PPARγ overexpression, and fabp4 was obviously increased through its overexpression (Fig. 6(c)). The DHA content was decreased by the suppression of PPARγ. However, the effect was restored by the overexpression of fabp4 (Fig. 6(d)). The observed changes in the other fatty acids are shown in online Supplementary Fig. S2.
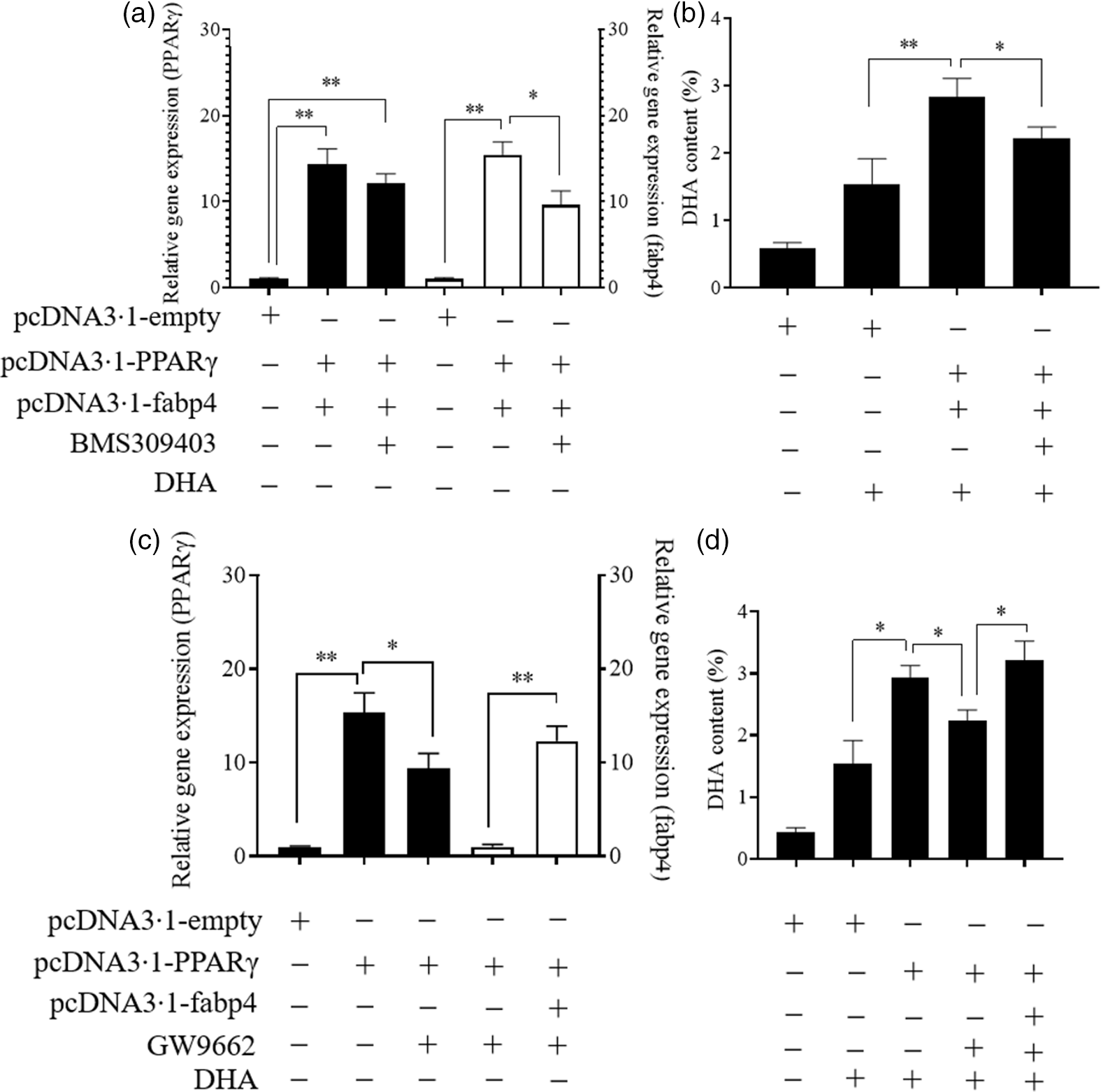
Fig. 6. PPARγ acts as a positive regulator in the fatty acid-binding protein 4 (fabp4)-mediated uptake of DHA. Values are expressed as means and standard deviations. *, P ≤ 0·05; **, P ≤ 0·01 (n 3). The Epinephelus coioides hepatocyte line (ECHL) cells were transfected with pcDNA3.1-empty alone or co-transfected with both pcDNA3.1-PPARγ and pcDNA3.1-fabp4. These plasmids were removed after 12 h and the cells were incubated with 75 μm BMS309403 and 50 μm DHA as shown for another 24 h (BMS309403 was added 2 h in advance). Cells were collected; the gene expression of PPARγ and fabp4 (a) and the content of DHA (b) were detected; The ECHL cells were transfected with pcDNA3.1-empty or pcDNA3.1-PPARγ for 12 h and then treated with 75 μm GW9662 for another 12 h without the plasmids. Afterward, pcDNA3.1-fabp4 was transfected into cells, and 50 μm DHA was added as shown to incubate the cells for another 12 h. Cells were collected to assess PPARγ and fabp4 mRNA levels (c) and DHA content was evaluated (d).
Discussion
In recent years, the n-3 LC-PUFA content in farmed fish has been redefined, resulting in fish being supplied with n-3 LC-PUFA levels that far exceed their biological requirements and with the aim of satisfying consumer demand(Reference Tocher2). Currently, this research area is receiving much focus, and two economic and effective approaches have been identified as potential avenues to increase the n-3 LC-PUFA content in farmed fish, namely the improvement of their biosynthesis ability and the enhancement of the uptake and deposition of dietary-limited n-3 LC-PUFA. Some regulators, such as microRNA, the hepatocyte nuclear factor 4α, and the sterol regulatory element-binding protein-1, have been demonstrated to be involved in the biosynthesis of n-3 LC-PUFA and can thus be used to further regulate the n-3 LC-PUFA content(Reference Dong, Wang and Chen24,Reference Ambros26,Reference Qin, Dalen and Gustafsson27) . In comparison, the uptake and deposition of n-3 LC-PUFA have received less attention. In our previous study, fabp4 was identified as a candidate gene involved in the uptake and deposition of n-3 LC-PUFA(Reference Lei, Li and Tian19). In the present study, we first cloned the promoter sequence of fabp4 in T. ovatus and then evaluated the serial deletion constructs of the fabp4 promoter in a dual-luciferase reporter assay, which showed that the core promoter was located between −2006 bp and −1521 bp upstream from the initiation codon. In mammals, the core promoter has generally been identified near the transcription initiation site(Reference Frith, Valen and Krogh28,Reference Megraw, Pereira and Jensen29) . For example, the core promoter of cattle (Bos taurus) fabp4 was present in the region of 272 bp upstream from the initiation codon(Reference Li, Zhao and Zhang30). The distinctive location of the T. ovatus fabp4 core promoter region indicated that there may be distinct regulatory mechanisms of fabp4 between T. ovatus and mammals. Alternatively, the transcription initiation site of T. ovatus fabp4 was far from the initiation codon; therefore, the core promoter was still near the transcription initiation site. This requires further investigation. In addition, the binding sites of transcription factors such as CCAAT/enhancer binding proteins, PPARγ, and Forkhead box O1 have been found in the core promoter region of T. ovatus fabp4, which is consistent with previous studies that showed that these transcription factors play a role in the transcriptional regulation of the fabp4 gene(Reference Li, Zhao and Zhang30–Reference Hua, Kim and Vo33).
Members of the FABP family, which consists of twelve members (FABP1–FABP12), are known to play important roles in fatty acid uptake(Reference Furuhashi and Hotamisligil34,Reference Ishimura, Furuhashi and Watanabe35) . In addition, a preference between the FABP subtype and fatty acid type has been suggested and is well studied in mammals(Reference Zimmerman, Moerkerk and Veerkamp36). In fish, studies have demonstrated that fabps are associated with dietary fatty acids and that a similar fatty acid-selective property has been conserved(Reference Torstensen, Nanton and Olsvik13,Reference Xu, Zhang and Wang14) , although the details regarding the FABP and its preferred fatty acid remain largely unclear. Recently, we identified fabp4 as a candidate fabp gene involved in n-3 LC-PUFA uptake and deposition(Reference Lei, Li and Tian19). In mammals, fabp4 is known to be involved in adipogenesis(Reference Samulin, Berget and Lien37,Reference Zhang, Zhao and Ning38) . In the present study, the overexpression of fabp4 triggered an increase in DHA content and the suppression of fabp4 alleviated this effect; this provides direct evidence that fabp4 contributes to the uptake and deposition of n-3 LC-PUFA. This result was similar to that of a previous study, in which the FABP4 protein from H. sapiens and M. musculus appeared to specifically bind to long-chain fatty acids (e.g. DHA) with high affinity(Reference Richieri, Ogata and Zimmerman39).
As early as 1996, the FABP family was reported to be regulated mainly at the transcriptional level(Reference Glatz and van der Vusse40). The transcription factor PPARγ is an important regulator for members of the FABP family both in mammals and in fish(Reference Mita, Beaulieu and Field41–Reference Laprairie, Denovanwright and Wright43), and the influence of PPARγ on fabp4 expression has been widely reported(Reference Hua, Kim and Vo33,Reference Pelton, Zhou and Demarest44–Reference Iso, Yajima and Kawaguchi46) . Analogously, in this study, the mutation of the PPARγ binding site significantly reduced the promoter activity of fabp4. Meanwhile, overexpression of PPARγ revealed a positive, dose-dependent effect on fabp4 promoter activity, indicating that the fabp4 gene was a target of PPARγ in T. ovatus.
PPARγ is believed to be a master regulator of adipogenesis(Reference Rosen and Spiegelman47). In C. idella, an increase in DHA content in the hepatopancreas is reportedly accompanied by the up-regulation of PPARγ (Reference Li, Liu and Ji48). Similarly, in this study, we found that PPARγ overexpression increased the DHA content and that the suppression of PPARγ mitigated this effect, thus indicating that PPARγ had a positive effect on DHA uptake and deposition. Our previous study indicated that PPARγ might be a key regulator of n-3 LC-PUFA uptake and deposition by modulating the genes (including fabp4) involved in fat emulsification and fatty acid transport(Reference Lei, Li and Tian19). In this study, the antagonist of fabp4, BMS309403 alleviated the increase in DHA content induced by PPARγ overexpression, indicating that fabp4 was required for DHA uptake and deposition evoked by PPARγ. Conversely, overexpression of fabp4 reversed the decline in the DHA content caused by GW9662 (a common antagonist of PPARγ). Collectively, the above findings demonstrate the functioning of fabp4 in the uptake and deposition on DHA in T. ovatus, and that such functions are regulated by PPARγ. Some natural ligands of PPARγ, such as α-linolenic acid and linoleic acid(Reference Edwards and O’Flaherty49,Reference Yu, Wu and Cheng50) , are common components of aquatic feeds. These ligands may further induce the expression of the PPARγ target gene fabp4, providing potential method to improve the n-3 LC-PUFA content of farmed fish in practical applications.
In the present study, we cloned the 5′ flanking sequence of T. ovatus fabp4 and revealed and characterised its important role in the regulation of n-3 LC-PUFA content in T. ovatus. Targeting fabp4 may, therefore, present an effective strategy for the regulation of the n-3 LC-PUFA content in this commercially important marine fish. Our findings also indicated that fabp4-mediated n-3 LC-PUFA uptake and deposition are probably regulated by PPARγ in T. ovatus. These results provide a new regulation axis that appears to be a key contributor to n-3 LC-PUFA content regulation in T. ovatus.
Acknowledgements
The authors wish to express their thanks to all the participants for their contribution to the present study. The authors are also grateful to the staff who supported this study in related research team. The following projects: Guangdong MEPP Fund (GDOE NO. 2019A30); China Agriculture Research System (CARS-47-G13); National Key R&D Program of China (2018YFD0900400) and the National Natural Science Foundation of China (No. 31873040; No. 42006099) provided the funding for this research.
The authors declare that there are no conflicts of interest.
Y. L., B. F. and C. L. conceived and designed the experiments. M. L. provided the experimental materials. Y. L., J. T. and C. L. co-wrote the paper.
Supplementary material
For supplementary material referred to in this article, please visit https://doi.org/10.1017/S0007114521000775








