


The development of adult CVD has been associated typically with classical risk factors such as poor diet, a sedentary lifestyle, smoking and genetic predisposition. However, the ‘Developmental Origins of Health and Disease’ (DOHaD) hypothesis identifies the uterine environment and associated maternal nutritional status as critical components in the origin of postnatal cardiovascular and metabolic disease(Reference Barker1–Reference McMillen and Robinson3). The DOHaD concept proposes that maternal nutritional status influences embryonic and fetal growth and physiology so that the setting of homeostatic mechanisms matches the nutrient availability in postnatal life. However, mismatch between pre- and postnatal nutrient environments is associated with adult onset disease(Reference Barker1–Reference McMillen and Robinson3).
Rodent and domestic animal studies have demonstrated that maternal gestational undernutrition can alter cardiovascular homeostasis in the offspring(Reference Brawley, Itoh and Torrens4–Reference Torrens, Brawley and Anthony8). The precise mechanisms underlying dietary programmed hypertension are likely to be multifactorial and complex, originating from more than one individual pathway. Vascular dysfunction in adult programmed animals in response to prolonged gestational undernutrition is characterised by an enhanced constrictive and impaired endothelial-dependent dilatation phenotype. In the sheep, maternal nutrient restriction encompassing either preconception or early gestation enhances vasoconstriction and attenuates vasodilatation in offspring coronary, thoracic and renal arteries(Reference Cleal, Poore and Boullin9–Reference Torrens, Snelling and Chau11). However, a more prolonged maternal gestational nutrition restriction associates with a higher offspring blood pressure and altered baroreflex responses to noradrenaline infusion(Reference Gopalakrishnan, Gardner and Rhind12). In the rat, male offspring of dams either globally undernourished (30 % less food than controls), or fed a low-protein diet (LPD), throughout pregnancy display significantly attenuated vascular responsiveness to both endothelial-independent and -dependent vasodilators(Reference Brawley, Itoh and Torrens4–Reference Torrens, Brawley and Anthony8, Reference Torrens, Hanson and Gluckman13). However, the magnitude of this vascular impairment was unaffected by a subsequent postnatal high-fat dietary challenge(Reference Torrens, Hanson and Gluckman13), highlighting the detrimental effect of poor maternal nutrition during pregnancy on vascular function.
Along with vascular function, homeostatic regulation of the renin–angiotensin system (RAS) also appears susceptible to programming in response to dietary and environmental challenges during gestation. The production of angiotensin II, a potent vasoconstrictor, from angiotensin I is mediated by angiotensin-converting enzyme (ACE)(Reference Skeggs, Kahn and Shumway14). The binding of angiotensin II to its receptor(Reference Higuchi, Ohtsu and Suzuki15) results in vasoconstriction, renal tubular Na reabsorption and aldosterone and vasopressin secretion(Reference Unger, Chung and Csikos16). Angiotensin II has also been implicated in endothelial dysfunction through its ability to increase oxidative stress(Reference Mehta and Griendling17). As a result, altered RAS regulation has been associated with the development of the hypertensive state(Reference Higuchi, Ohtsu and Suzuki15) and with several cardiovascular and renal pathologies(Reference Lucius, Gallinat and Busche18, Reference Unger19). The role of RAS in the aetiology of programmed adult hypertension following maternal nutrient restriction has been demonstrated via the normalisation of blood pressure in hypertensive adult offspring from LPD-fed rat dams after administration of the ACE inhibitor, captopril(Reference Langley-Evans and Jackson20), or through the blockade of the angiotensin I receptor (AT1R) with losartan(Reference Sherman and Langley-Evans21). Offspring from rat dams fed LPD during gestation display significantly altered mRNA and protein levels for the angiotensin I and II receptors (AT1R and AT2R) in kidney tissue(Reference McMullen, Gardner and Langley-Evans22, Reference Vehaskari, Stewart and Lafont23). Maternal LPD has also been shown to alter expression levels of the glucocorticoid receptor and the Na/K-adenosine triphosphatase subunits in the kidney(Reference Bertram, Trowern and Copin24). These findings have led to the suggestion that maternal undernutrition could alter renal Na retention, contributing to the increased systolic blood pressure (SBP) observed in these animals.
The pre-implantation period of development has been shown to be particularly sensitive to changes in its immediate environment, whether in vivo or in vitro, resulting in a myriad of changes in offspring phenotype(Reference Fernandez-Gonzalez, Moreira and Bilbao25–Reference Watkins, Ursell and Panton29). Altered cardiovascular and metabolic phenotype, including raised SBP and diastolic blood pressure and altered glucose homeostasis, have been identified in human in vitro fertilisation children compared with age-matched controls(Reference Ceelen, van Weissenbruch and Vermeiden30). In the sheep, culture of pre-implantation embryos in the presence of serum resulted in significantly altered rates of blastocyst development, and elevated growth of placental and fetal tissues(Reference Rooke, McEvoy and Ashworth31). Impaired blastocyst and fetal development have also been reported following the culture of mouse pre-implantation embryos under high ammonia(Reference Zander, Thompson and Lane32) or reduced oxygen(Reference Feil, Lane and Roberts33) environments. Studies from our laboratory have demonstrated that maternal LPD diet targeted exclusively to the pre-implantation period in rodents significantly alters blastocyst cell numbers and gene expression patterns in fetal liver tissue(Reference Kwong, Wild and Roberts26, Reference Watkins, Ursell and Panton29, Reference Kwong, Miller and Ursell34, Reference Kwong, Miller and Wilkins35), whilst elevating offspring postnatal growth, behavioural patterns and mean lifelong SBP(Reference Watkins, Ursell and Panton29).
Whilst the development of offspring hypertension following maternal gestational undernutrition is well established, the mechanisms and pathways involved are still poorly defined. In the present study we evaluate for the first time key mechanisms of adult blood pressure regulation, in order to gain a better understanding of how maternal undernutrition during discrete windows of gestation can programme disease in the adult offspring.
Methods
Animal treatments
All experiments were conducted using protocols approved by the UK Home Office and local ethics committee. Female MF-1 mice were bred in-house (University of Southampton Biomedical Research Facility) and maintained on a 07.00 to 19.00 hours light cycle with controlled temperature and standard chow and water ad libitum. All offspring and archived tissues investigated in the present study were taken from the same cohort of animals as described previously(Reference Watkins, Ursell and Panton29). Briefly, virgin MF-1 females (aged 7–8·5 weeks) were mated naturally with MF-1 studs (Harlan Link Ltd, Bicester, Oxon, UK) and randomly assigned to either (a) a normal protein diet (NPD; 18 % casein) throughout gestation, (b) an isocaloric low-protein diet (LPD; 9 % casein) throughout gestation, or (c) the LPD for the first 3·5 d before switching to the NPD for the remainder of gestation (Emb-LPD). The morning after mating, females were individually coded and fed such that the experimenter was blinded to the diet each female received, and subsequently did not know what dietary treatment any offspring pertained to. Nineteen litters of each treatment group were generated. Mothers from the day of birth, and offspring (after weaning at 3 weeks), were fed standard chow. Litter size was adjusted to six at birth (three per sex) and the sexes caged separately from weaning. Previously, we demonstrated that these dietary regimens were sufficient to induce mean lifelong hypertension in both male (elevated by 3·2 mmHg) and female (elevated by 3·0 mmHg) offspring as assessed by tail-cuff plethysmography(Reference Watkins, Ursell and Panton29), and that female Emb-LPD offspring displayed increased body weight from birth for up to 28 weeks (increased by 11 % at birth, and 8·3 % at 28 weeks, respectively, over controls(Reference Watkins, Ursell and Panton29). Analysis of the mean lifelong SBP (measurements taken at 9, 15 and 21 weeks of age) of the cohort of males and females used within all experiments within the present study (fifty-five, fifty-seven and sixty-one NPD, Emb-LPD and LPD offspring, respectively) revealed that both Emb-LPD and LPD offspring had significantly elevated mean lifelong SBP when compared with the control offspring used (NPD 103·93 (sem 0·61) mmHg; Emb-LPD 107·46 (sem 0·43) mmHg; LPD 107·3 (sem 0·53) mmHg; P < 0·001). Therefore, the mice in the cohort analysed in the present study were considered as being representative of the entire population(Reference Watkins, Ursell and Panton29) from which they were selected.
Mesenteric artery vasoreactivity
Vascular function in male offspring was assessed at 22 weeks in isolated small mesenteric artery segments following cervical dislocation as described previously(Reference Torrens, Brawley and Barker7, Reference Torrens, Brawley and Anthony8, Reference Torrens, Hanson and Gluckman13, Reference Watkins, Wilkins and Cunningham36). Male offspring alone were analysed in order to minimise potential variation caused from the differing levels of circulating oestrogen in female offspring during the oestrous cycle, and the effect this has on vascular responsiveness(Reference Liu, Hattori and Fukao37). Briefly, small mesenteric artery segments (luminal diameter about 100–300 μm) were isolated and mounted on a wire myograph (Danish Myo Technology A/S, Aarhus, Denmark). Following normalisation, cumulative concentration response curves were measured for the α1-adrenergic agonist phenylephrine (10− 9 to 10− 4 mol/l). After pre-constriction with a submaximal dose of phenylephrine (80 % of maximal excitatory concentration; EC80), cumulative concentration response curves to the endothelial-dependent and -independent vasodilators acetylcholine (10− 9 to 10− 5 mol/l) and isoprenaline (10− 10 to 10− 6 mol/l), respectively, and the NO donor, sodium nitroprusside (10− 11 to 10− 5 mol/l), were conducted in that order in the same mesenteric arteries from seven or eight individual males, all from different litters. All drugs were purchased from Sigma (Poole, Dorset, UK).
Kidney glomerular counting
Due to the fact that male offspring alone had been analysed for the study of mesenteric artery responsiveness (for reasons given above), only female kidneys were analysed in order to maintain a balanced number of male and female offspring for the final analysis of organ allometry at 28 weeks (organ allometry data previously detailed by Watkins et al. (Reference Watkins, Ursell and Panton29)). This also kept the present study in line with our previous study(Reference Watkins, Wilkins and Cunningham36), making it possible to compare results. Stereological analysis of kidney glomerular number was conducted as previously described(Reference Watkins, Wilkins and Cunningham36). Briefly, left kidneys from female offspring were perfusion-fixed with neutral buffered formalin (BDH Laboratory Supplies, Poole, Dorset, UK). Kidney volume was determined by suspended immersion into neutral buffered formalin placed on a balance, with the increase in weight recorded being equal to the volume of the kidney. Three separate measurements of volume per kidney were taken and an average calculated. Fixed kidneys were then processed into glycol methacrylate (JB-4 Embedding Kit; Polysciences Europe GmbH, Eppelheim, Germany) and exhaustively sectioned at 2 μm. Every 100th and 110th section was analysed using the Stereo Investigator software (Microbrightfield, Williston, VT, USA). An unbiased counting frame (500 μm2) was placed randomly over the 100th section; glomeruli present within the 110th but absent from the 100th section were counted and an estimate of kidney glomerular number was obtained using the fractionator principle(Reference Bertram38).
RNA extraction and quantitative RT-PCR for gene expression analysis
Whole left kidneys from eight male and eight female offspring per treatment group at 28 weeks, archived from our previous study(Reference Watkins, Ursell and Panton29), were powdered and the RNA extracted using the RNeasy® Lipid Tissue Mini Kit (QIAGEN, Crawley, West Sussex, UK) according to the manufacturer's instructions. On-column DNase I digestion was performed to remove contaminating genomic DNA. RNA was reverse transcribed to cDNA using the ImProm™II kit (Promega, Southampton, Hants, UK), according to the manufacturer's instructions. Intron-spanning primers used in the present study are detailed in Table 1. For real-time PCR, a mastermix was prepared consisting of 10 μl 2X Precision Mastermix containing SYBRgreen (PrimerDesign, Southampton, Hants, UK), 1·2 μl primer mix (containing 5 μm each forward and reverse primers) and 7·8 μl water per reaction. Mastermix was aliquotted to ninety-six-well PCR plates (Axygen, Union City, CA, USA) in 19 μl volumes and 5 ng cDNA was added to sample wells, or water to control (no template) wells, giving a final reaction volume of 20 μl. All samples were run in triplicate. An inter-plate calibrator was included on all runs to control for inter-assay variation. Amplification and detection were performed using a DNA Engine thermal cycler and Chromo4 Real-Time Detector and data were acquired using Opticon Monitor version 3.1 software (Bio-Rad, Hemel Hempstead, Herts, UK). A post-amplification melting curve confirmed the presence of specific products for each primer set. Data were normalised to the expression of succinyl dehydrogenase α (Sdha) and phosphoglycerate kinase 1 (Pgk1) using geNorm software (Ghent University, Ghent, Belgium)(Reference Vandesompele, De Preter and Pattyn39), with which we determined these to be the two most stable reference genes in adult mouse kidney (data not shown).
Table 1 Real-time PCR primer details for gene expression studies

Angiotensin-converting enzyme activity
Serum and lung ACE activity was analysed as previously described(Reference Watkins, Platt and Papenbrock28, Reference Watkins, Wilkins and Osmond40). Briefly, samples were incubated in the presence of hippuryl-l-histidine-l-leucine acetate salt (Sigma, Poole, Dorset, UK) solution in buffer at 37°C followed by the addition of cyanuric chloride (Sigma) in 1,4 dioxane (Sigma). Each sample was analysed in duplicate, with four readings per duplicate analysed at 380 nm. Samples containing only serum or lung lysate and buffer were used as negative controls. Sample readings were analysed against a standard curve ranging from 50 μm to 1 mm, prepared in buffer and treated exactly as the samples. Serum and lung ACE activity was expressed as nmol hippurate formed per ml serum per min or per mg protein per min, respectively.
Statistical analysis
For analysis of vascular responsiveness data, a four-parameter logistic equation using non-linear regression was used to obtain the − log effective concentration equal to 50 % of the maximal response (pEC50) and maximum response for each of the cumulative concentration response curves analysed. Differences in maximal response and pEC50 were analysed using two-way ANOVA (Prism 3·0, GraphPad Software Inc., San Diego, CA, USA) with Bonferroni post hoc correction as described previously(Reference Torrens, Brawley and Barker7, Reference Torrens, Hanson and Gluckman13, Reference Watkins, Wilkins and Cunningham36). Kidney glomerular number and gene transcript analysis and ACE data were analysed using a multilevel random effects regression model (SPSS version 15; SPSS, Inc., Chicago, IL, USA) taking into account the hierarchical nature of the data with between-mother and within-mother variation and different parameters measured from individual animals(Reference Watkins, Platt and Papenbrock28, Reference Watkins, Ursell and Panton29, Reference Kwong, Osmond and Fleming41). Therefore, statistical differences identified between treatment groups were independent of maternal origin of litter, gestational litter size and body weight, unless otherwise stated. All data are presented as mean values with their standard errors of the mean.
Results
Mesenteric artery vasoreactivity
Isolated male arteries from all treatment groups displayed a concentration-dependent vasoconstriction to the α1-adrenergic agonist phenylephrine, which was not different between treatments (Fig. 1(a)). Following pre-constriction with phenylephrine, no difference in responsiveness to the endothelium-dependent vasodilator acetylcholine was observed between treatment groups (Fig. 1(b)). However, Emb-LPD males displayed an attenuated response to the β-adrenoceptor agonist isoprenaline when compared with NPD arteries (% maximal response 62·22 (sem 4·59) v. 76·11 (sem 3·58), respectively; P = 0·025) whilst the response in LPD arteries was shifted significantly to the right (significantly different pEC50) when compared with NPD arteries (pEC50, 6·99 (sem 0·07) and 7·56 (sem 0·13), respectively; P = 0·04; Fig. 1(c)), both of which were independent of litter size and body weight effects. No differences in response to sodium nitroprusside were observed between the treatment groups.
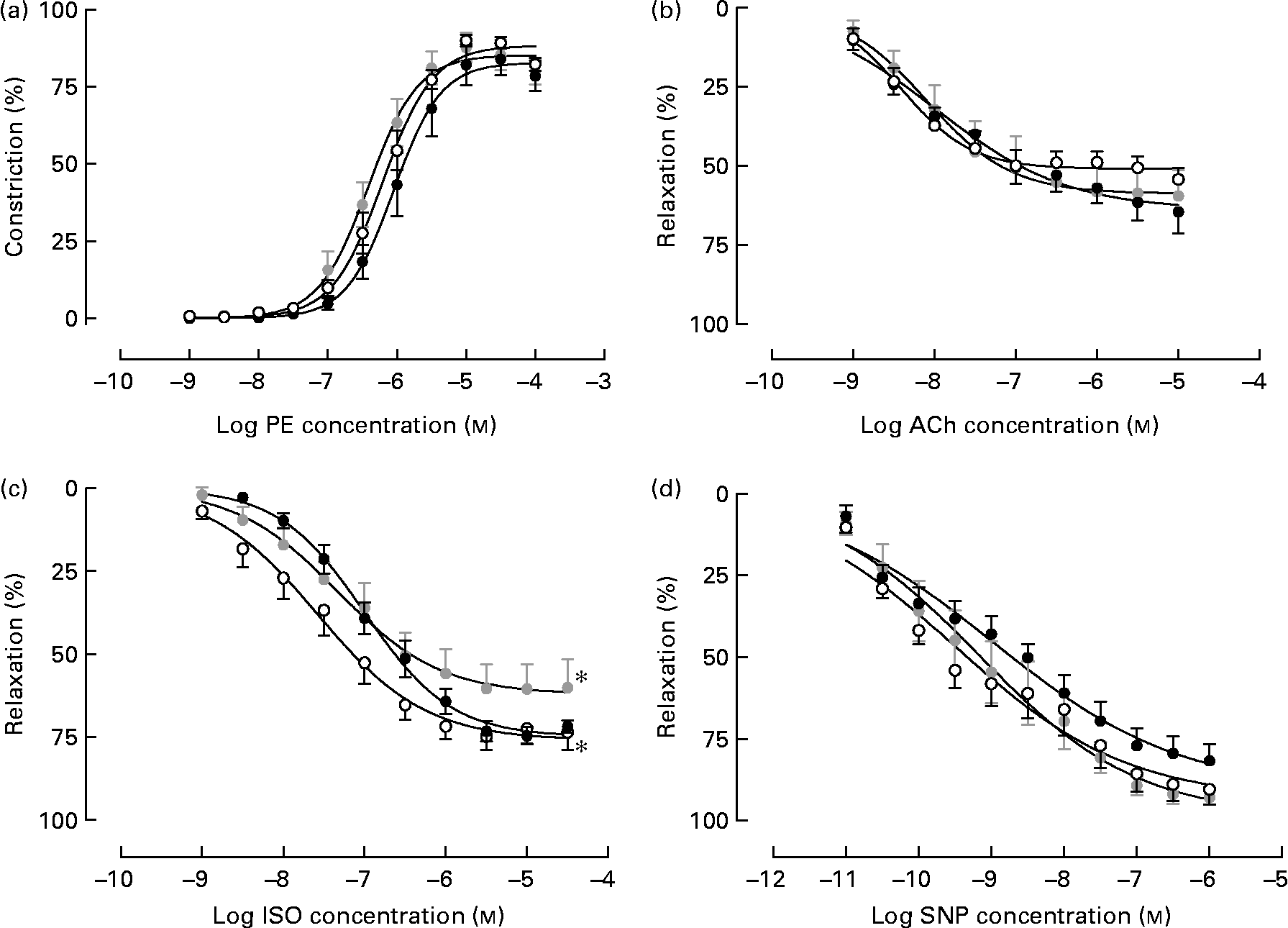
Fig. 1 Vasoreactivity of isolated mesenteric arteries from male offspring at 22 weeks of age from dams fed a low-protein diet throughout gestation (LPD; ●) or exclusively during the pre-implantation period (Emb-LPD; ![]() ) compared with arteries of offspring of normal-protein diet (NPD)-fed dams (○). Cumulative additions of (a) phenylephrine (PE) and, after pre-constriction with PE, of the vasodilators (b) acetylcholine (ACh), (c) isoprenaline (ISO) and (d) sodium nitroprusside (SNP). Values are means of seven to eight males, each from different litters representing each treatment group, with standard errors represented by vertical bars. * Response was significantly different from that of the NPD arteries (P < 0·05).
) compared with arteries of offspring of normal-protein diet (NPD)-fed dams (○). Cumulative additions of (a) phenylephrine (PE) and, after pre-constriction with PE, of the vasodilators (b) acetylcholine (ACh), (c) isoprenaline (ISO) and (d) sodium nitroprusside (SNP). Values are means of seven to eight males, each from different litters representing each treatment group, with standard errors represented by vertical bars. * Response was significantly different from that of the NPD arteries (P < 0·05).
Kidney glomerular counts
Stereological analysis of female left kidneys at 28 weeks revealed no difference in mean glomerular number between the treatment groups, or in mean kidney weight or mean kidney volume (Fig. 2).
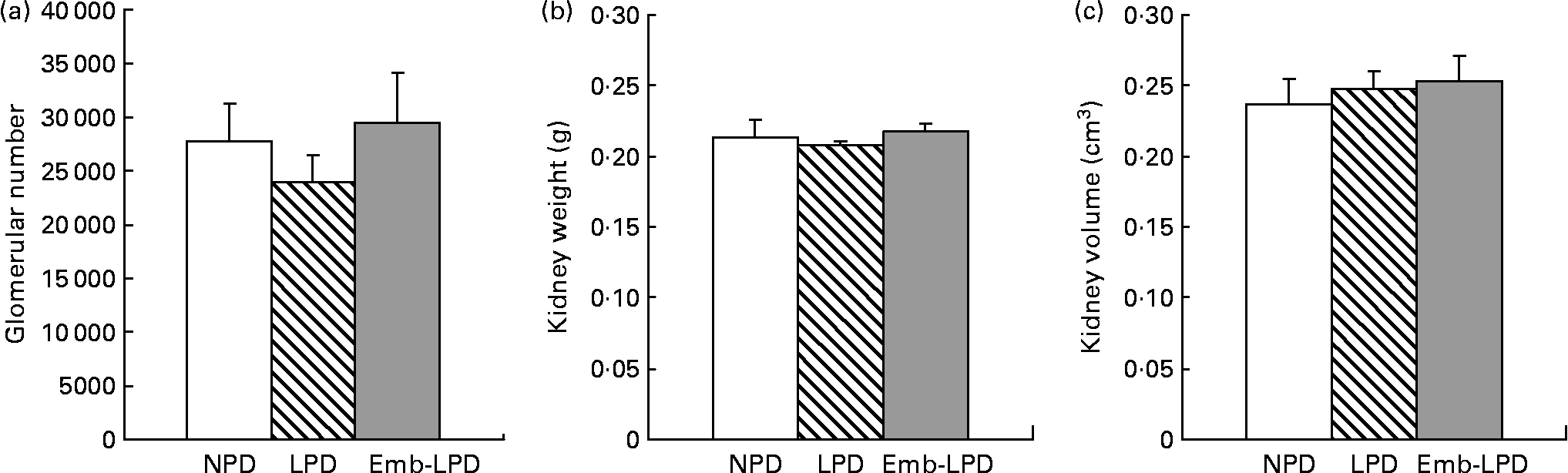
Fig. 2 Effect of maternal low-protein diet either throughout gestation (LPD) or exclusively during the pre-implantation period (Emb-LPD) compared with maternal normal-protein diet (NPD) on kidney glomerular number (a), kidney weight (b) and kidney volume (c) in female offspring at 28 weeks. Values are means for six kidneys per treatment, each from different litters representing each treatment group, with standard errors represented by vertical bars.
Kidney gene expression analysis
Real-time analysis of type 1a angiotensin II receptor (Agtr1A), the α1, β1, 2 and 3 subunits of the Na+/K+ ATPase transporter (Atp1a1, Atp1b1, Atp1b2, Atp1b3) and glucocorticoid receptor (Nr3c1) mRNA expression in male and female left kidneys revealed no significant differences between any of the treatment groups (Table 2).
Table 2 Real-time RT-PCR relative expression values from male and female kidney tissue*
(Mean values with their standard errors)
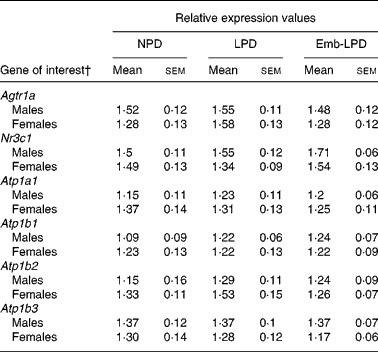
NPD, normal-protein diet; LPD, isocaloric low-protein diet; Emb-LPD, low-protein diet for the first 3·5 d before switching to normal diet for the remainder of gestation.
* Eight males and eight females, each from different litters representing each treatment group.
† For gene names, see Table 1.
Serum and lung angiotensin-converting enzyme activity
Analysis of serum ACE activity showed no differences between male treatment groups; however, LPD females had an elevated activity compared with NPD females (135·45 (sem 11·65) v. 113·17 (sem 9·48) nmol hippurate formed/ml serum per min, respectively; Fig. 3(a); P = 0·044). Analysis of lung ACE activity showed that Emb-LPD males had an elevated activity when compared with NPD and LPD males (3·98 (sem 0·48) v. 1·69 (sem 0·16) and 2·33 (sem 0·29) nmol hippurate formed/mg protein per min, respectively; Fig. 3(b); P ≤ 0·005). Whilst Emb-LPD females had an elevated activity when compared with LPD females (3·31 (sem 0·55) v. 2·29 (sem 0·41) nmol hippurate formed/mg protein per min, respectively; Fig. 3(b); P = 0·05), there was no difference between Emb-LPD females and LPD and control females. All differences were independent of litter size effects; however, body weight at 28 weeks of age had a negative influence on female lung ACE activity ( − 0·104 nmol hippurate formed/mg protein per min per g increase in body weight; P = 0·018).
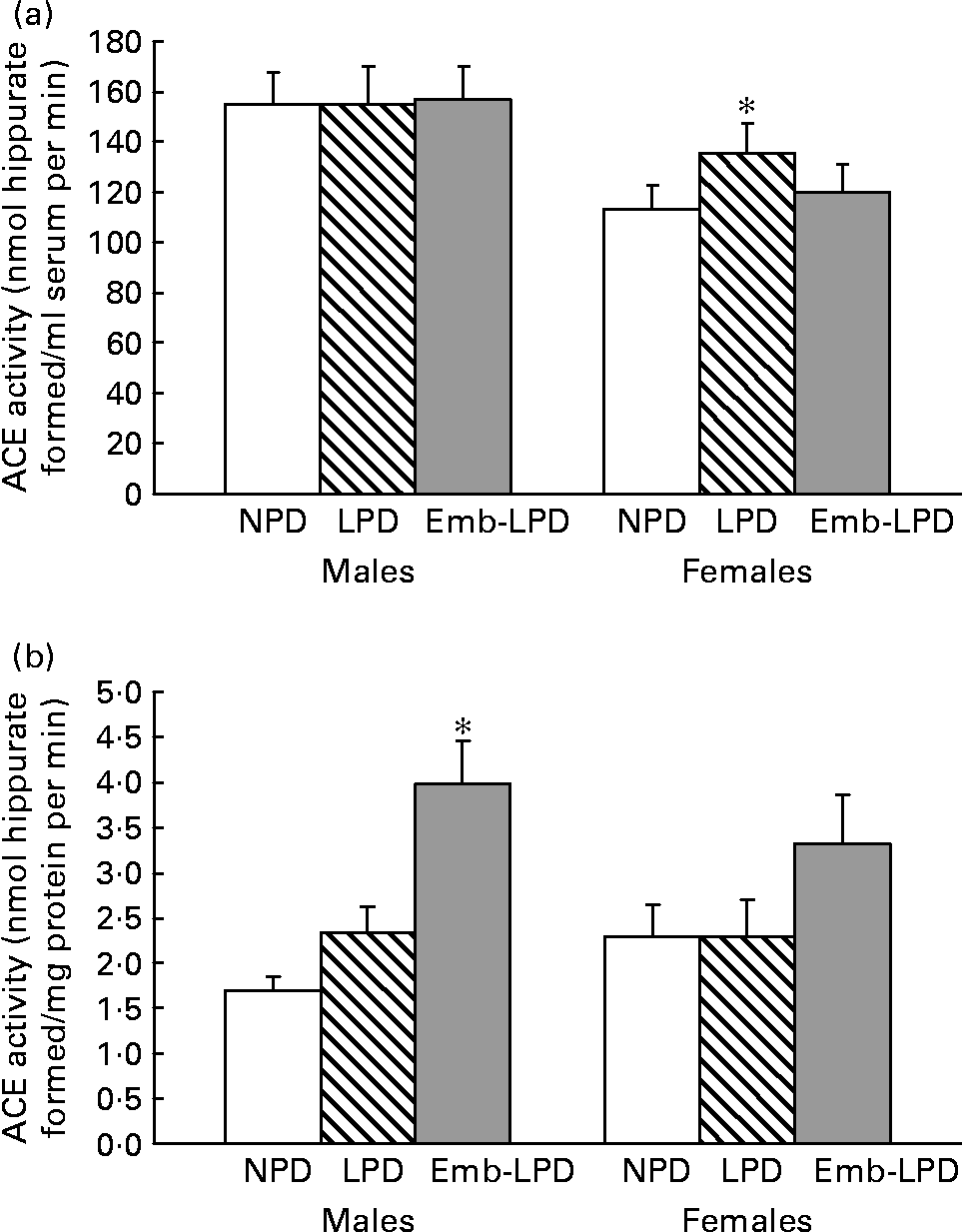
Fig. 3 Enzymic activity of serum (a) and lung tissue (b) angiotensin-converting enzyme (ACE) activity in male and female offspring at 28 weeks. NPD, normal-protein diet; LPD, low-protein diet; Emb-LPD, low-protein diet for the first 3·5 d before switching to normal diet for the remainder of gestation. Values are means for seventeen to nineteen males and females per treatment, each from different litters representing each treatment group, with standard errors represented by vertical bars. * Mean value was significantly different from that of the NPD group (P < 0·05).
Discussion
The earliest stages of development have been highlighted as critically sensitive periods during which perturbations, either in vivo or in vitro, can have long-term effects on offspring health and physiology(Reference Fernandez-Gonzalez, Moreira and Bilbao25–Reference Watkins, Ursell and Panton29, Reference Rooke, McEvoy and Ashworth31, Reference Feil, Lane and Roberts33, Reference Watkins, Wilkins and Cunningham36). Previously, we have demonstrated that a maternal LPD treatment exclusively during the pre-implantation period (the first 3·5 d of gestation; Emb-LPD) or throughout gestation (LPD) significantly elevated offspring SBP(Reference Watkins, Ursell and Panton29). Therefore, the present study investigated the mechanisms underlying the observed elevated SBP in a cohort of these same offspring, focusing on vascular function, the RAS and changes in nephron number and kidney gene expression patterns. We observed impaired vasodilatation in male LPD and Emb-LPD offspring, elevated patterns of serum and lung ACE activity in LPD females and Emb-LPD males, respectively, but no change in kidney glomerular number in females or kidney gene expression in male and female offspring. These data further strengthen the association between the predisposition to chronic adult-onset diseases and an impaired uterine environment during critical stages of gestation and especially before implantation.
Our first major finding was that isolated mesenteric arteries from Emb-LPD and LPD male offspring displayed significantly attenuated responsiveness to the β-adrenoceptor agonist, isoprenaline. The association of CVD and attenuated vascular responsiveness has been demonstrated previously in offspring from LPD fed rats(Reference Brawley, Itoh and Torrens4, Reference Torrens, Brawley and Barker7, Reference Torrens, Brawley and Anthony8, Reference Lamireau, Nuyt and Hou42) and in offspring from dietary induced obese mice(Reference Samuelsson, Matthews and Argenton43). The present data, however, identify the importance of nutrition during the pre-implantation period in the maintenance of vascular tone. Since endothelial-dependent vasodilatation appeared unaffected, as gauged by similar responses to acetylcholine between treatment groups, it might suggest that the vascular defect lies in altered smooth muscle signalling via cAMP, rather than the bioavailability of responsiveness to NO. Isoprenaline produces vasodilatation through activation of cAMP(Reference Tang and Gilman44); however, in mouse mesenteric arteries this is dependent on a functioning endothelial NO synthase(Reference Longo, Jain and Vedernikov45). One suggestion is that basal endothelial NO release can modulate isoprenaline responses by inhibiting phosphodiesterase breakdown of cAMP(Reference Delpy, Coste and Gouville46). From the present study the role of NO is unclear, as responses were not performed in the presence of an NO synthase inhibitor such as N-nitro-l-arginine-methyl ester. Furthermore, reductions in the expression of guanylate cyclase or intracellular cGMP concentrations have been demonstrated in models of impaired endothelium-independent responsiveness(Reference Kagota, Tamashiro and Yamaguchi47–Reference Ruetten, Zabel and Linz49). It is therefore possible that alterations in guanylate cyclase or cGMP may be contributing to the blunted vasodilatation responses observed in the male offspring within the present study; however, this would require further investigation to confirm. Similarly, whether differences in vasodilatation exist for female offspring from LPD and Emb-LPD mothers also remains to be determined. However, vascular responsiveness at different stages of the oestrous cycle would have to be determined so that both dietary and hormonal effects could be quantified independently.
The second major alteration in postnatal phenotype that we observed was that female LPD offspring displayed significantly elevated serum ACE activity, whilst Emb-LPD males displayed elevated lung ACE activity. The association between ACE and SBP is well documented. ACE− / − mice are hypotensive, with a mean SBP about 30 mmHg lower than wild-type mice(Reference Esther, Marino and Howard50, Reference Klein, Le Quach and Cole51). Hypertensive offspring from LPD-fed rat dams show a normalisation in their SBP after administration of the ACE inhibitor captopril(Reference Langley-Evans and Jackson20) whilst hypertensive mice developing from embryos genetically programmed to develop at a slower rate, or from in vitro cultured embryos, display significantly elevated serum ACE activities(Reference Watkins, Platt and Papenbrock28, Reference Watkins, Wilkins and Osmond40). In human subjects, serum ACE levels are significantly higher in hypertensive as opposed to normotensive subjects(Reference Forrester, McFarlane-Anderson and Bennett52), with serum ACE levels correlating positively with plasma angiotensin II levels(Reference Nystrom, Karlberg and Ohman53). The programming of elevated serum and tissue ACE activity has also been demonstrated following maternal cortisol infusion(Reference Forhead, Broughton Pipkin and Fowden54), and co-occurring with adult elevated SBP in response to pre-implantation embryo culture conditions(Reference Watkins, Platt and Papenbrock28) and maternal diet(Reference Watkins, Wilkins and Cunningham36). As in our previous studies(Reference Watkins, Platt and Papenbrock28, Reference Watkins, Wilkins and Osmond40) we observed sex- and tissue-specific responses in ACE activity across treatment groups. In vivo, serum ACE is derived from the cleavage of endothelial-bound somatic ACE from the lungs, vasculature and kidneys. As we only assessed the activity of ACE in the serum and lungs, we are not able to comment on the activity of other somatic sources of ACE, their relative contribution to the overall activity in the serum or in possible differences between the sexes.
The third finding was that female left kidney glomerular number was unaltered in response to maternal dietary protein levels. Significant reductions in both kidney size and glomerular number have been reported in response to maternal LPD in the rat(Reference Langley-Evans, Welham and Jackson55), mouse(Reference Hoppe, Evans and Bertram56) and with decreased birth weight in humans(Reference Manalich, Reyes and Herrera57). However, several studies have demonstrated programmed elevated blood pressure in the absence of significant changes in glomerular number(Reference Ortiz, Quan and Zarzar58–Reference Woods, Ingelfinger and Rasch60). Whilst reductions in glomerular number may reflect compromised kidney development during gestation in females, in response to restricted maternal nutrition, nephrogenesis in the mouse and rat is not complete until several days after birth(Reference Moritz and Wintour61). As all dietary challenges ceased by time of birth any reduction in glomerular number occurring during gestation in response to the LPD could potentially be compensated for during early postnatal life. This may be one reason for the similar glomerular numbers, kidney weights and volumes observed in the present study in the female offspring. In the present study, only female offspring were analysed and so it would also be of interest to determine whether glomerular number was similarly unaffected in males.
Examination of gene expression in the left kidneys of male and female offspring found no differences in the expression of glucocorticoid receptor transcripts or in any of the Na+/K+ ATPase subunits examined. This is in contrast to studies in the rat model, whereby exposure to maternal LPD has led to reported 2-fold increases in glucocorticoid receptor mRNA expression, as well as in transcripts for the α1 and β1 subunits of the Na+/K+ ATPase(Reference Bertram, Trowern and Copin24), in association with raised SBP in the order of 20–30 mmHg. The present study also found no differences in expression of the angiotensin II type 1A receptor (Agtr1a) between the kidneys of treated and control offspring. In the rat model, while up-regulated Agtr1a mRNA has been observed in LPD offspring from one study at 4 weeks of age(Reference Vehaskari, Stewart and Lafont23), another study reported no difference in expression of this transcript(Reference McMullen, Gardner and Langley-Evans22). This may reflect differences in the timing and severity of the dietary manipulation between these two studies, whereby the altered Agtr1a expression was observed after exposure of dams to a 6 % casein diet from day 12 of pregnancy(Reference Vehaskari, Stewart and Lafont23) but not after exposure to a 9 % diet from the day of mating until delivery(Reference McMullen, Gardner and Langley-Evans22). This discrepancy reinforces the hypothesis that the specific mechanistic origins of the programmed hypertension phenotype are driven by the timing and severity of the nutritional challenge.
It is of interest to compare the relative difference in postnatal phenotype observed in the present study with those occurring in response to maternal LPD during oocyte maturation(Reference Sherman and Langley-Evans21). During the present study, we observed endothelial-independent impairments in vasodilatation, elevated ACE activity profiles, but no changes in glomerular number. However, in response to the LPD during oocyte maturation, we observed impaired endothelial-dependent and -independent vasodilatation, no changes in ACE activity and an elevation in glomerular number(Reference Watkins, Wilkins and Cunningham36). As previously discussed in detail(Reference Watkins, Wilkins and Cunningham36), these changes may reflect the different times during the reproductive cycle during which the LPD was administered (gamete maturation v. post-fertilisation development). Alternatively, they may reflect the contribution of the paternal genome, absent during oocyte maturation(Reference Angiolini, Fowden and Coan62, Reference Smith, Garfield and Ward63). A third distinction is the connection between early postnatal phenotype and adult physiology and cardiovascular health present in this and previous associated studies(Reference Watkins, Ursell and Panton29) which appear absent in the offspring derived from the LPD administered during oocyte maturation(Reference Watkins, Wilkins and Cunningham36). Whilst both dietary protocols result in hypertensive offspring, the mechanisms underlying this state appear both similar (vascular responsiveness) and distinct (ACE activity, body weight and kidney morphology).
In conclusion, we have demonstrated that maternal LPD given either exclusively during pre-implantation embryo development, or throughout gestation, results in an impaired arterial vasodilatory capacity in male offspring, and altered activity of serum and lung ACE activity in female and male offspring, respectively. However, kidney glomerular number in females, and expression of key RAS component transcripts and blood volume regulators in both male and female offspring were unaffected. These findings demonstrate that maternal undernutrition encompassing the earliest stages of development has an impact upon interacting vascular, signalling and enzymic pathways which associate with the development of an elevated offspring SBP phenotype(Reference Watkins, Ursell and Panton29). These changes in offspring homeostatic regulation may have occurred in response to adaptations made during embryonic and/or fetal development to maximise postnatal development in an anticipated nutritional environment, but which then predispose the offspring to adult disease. Collectively, the present results offer novel insights into the mechanisms behind the association between poor pre-implantation maternal diet and adult cardiovascular dysfunction.
Acknowledgements
The present study was supported by the Biotechnology and Biological Sciences Research Council (BBSRC; grant no. BBF007450), the National Institutes of Health, USA as part of the National Institute of Child Health and Human Development (NICHD) National Cooperative Program on Female Health and Egg Quality under cooperative agreement (grant no. U01 HD04435) and the Gerald Kerkut Charitable Trust. M. A. H. is supported by the British Heart Foundation. We thank the Biomedical Facility, University of Southampton, for their technical support. We also acknowledge the help of Stephanie Marfy-Smith with the preparation of the kidney tissue for gene expression analyses.
A. J. W., E. S. L., C. T., J. K. C., W. P. G., J. J. E. and T. P. F. designed the research; A. J. W., E. S. L. and L. G. performed the research; A. J. W., E. S. L., C. T. and C. O. analysed the data; A. J. W., E. S. L., C. T., M. A. H. and T. P. F. wrote the manuscript.
There are no conflicts of interest associated with the present study.





