


In 2002, a joint FAO/WHO Working Group defined a probiotic as a ‘live microorganism which when administered in adequate amounts confers a health benefit on the host’(1). Until now, specific probiotics have been selected and characterised based on their in vitro properties, for example tolerance to gastrointestinal conditions, adhesion to the intestinal mucus or epithelial cells and competitive exclusion of target pathogens(Reference Tuomola, Crittenden and Playne2–Reference Collado, Meriluoto and Salminen4). Unfortunately, published information frequently lacks accurate information on strain identity and characteristics, for example International Culture Collection deposits and deposit numbers, on the one hand, and the mode of administration and the preclinical documentation and the selection criteria on the other. This makes strain comparison difficult and may influence the results of clinical intervention studies. We have extended this concern to the food matrix used to deliver the probiotic to the study subject, providing experimental evidence that the impact of Lactobacillus rhamnosus GG (ATCC 53103) on antigen transport depends on the quality of protein, unhydrolysed v. hydrolysed(Reference Pessi, Sutas and Marttinen5) in the diet.
One further cause of variation in the outcomes of clinical studies, beyond differences in populations, selection criteria and study design, may arise when different production conditions, growth media, drying conditions or cryoprotectants are used for the same strain or when a successful probiotic is combined with other bacteria or strains. For example, the adhesion properties of the L. rhamnosus GG strain (ATCC 53103) have been shown to depend on the composition of the growth media and the number of starter culture transfers(Reference Elo, Saxelin and Salminen6). Clinical and immunological effects were not achieved when L. rhamnosus GG was combined with L. rhamnosus LC705, Bifidobacterium breve Bb99 and Propionibacterium freudenreichii ssp. shermanii(Reference Kukkonen, Savilahti and Haahtela7). Similarly, as far back as in 1983, a distinction in the clinical outcome for L. acidophilus was shown to depend on the production lot(Reference Clements, Levine and Ristaino8).
The use of probiotics is currently extending from research to recommendations(Reference Guarino, Albano and Ashkenazi9, Reference Floch, Walker and Guandalini10), but rigorous scientific effort is still required to validate specific strains with anti-allergic potential for preventive and therapeutic applications. The production of functional probiotic foods presupposes stability of strain features in the final product. This requirement is emphasised in treating young infants with impaired gut barrier function, aberrant gut microbiota and sensitisation to dietary substances. For this purpose, we evaluated in the present study the strain characteristics, tolerance to acid pH and the adhesion and competitive exclusion properties of putative L. rhamnosus GG isolates obtained from different commercial products and production lots used in clinical intervention studies. As a prerequisite step, each isolate was genotypically and phenotypically characterised to ensure identical profiles, also excluding the possibility of contamination or misidentification of the strain obtained from different probiotic products.
We hypothesised that there are differences among the properties of the same probiotic strain, which depend on the production processes using different methods and media, and these influence strain properties with a consequent impact on the clinical and nutritional properties of probiotics.
Experimental methods
Bacterial isolates and culture conditions
Altogether thirteen L. rhamnosus isolates claimed to be L. rhamnosus GG (ATCC 53103) were isolated from different probiotic products from different countries (identification codes: AL, BL, CL, DL, EL, FL, HL, IL, JL, KL, LL, ML and NL). Among them, four isolates (AL, EL, FL and NL) were isolated from capsule products from different countries, two isolates (LL and ML) were derived from commercial infant foods, three isolates (DL, HL and KL) were isolated from freeze-dried powders of different products and four isolates (BL, CL, IL and JL) were provided from soft agar. For comparison, the original L. rhamnosus GG (original L. rhamnosus GG strain isolate donated by Professor Sherwood Gorbach of Tufts University, Boston, MA, USA) was included. The original L. rhamnosus GG strain is the foundation strain from which other cultures used in probiotic products were derived. An additional commercial probiotic L. rhamnosus strain (identification code OL) of the same species but known to be phenotypically different from L. rhamnosus GG was included as an external reference strain.
The bacterial pathogens used were Cronobacter sakazakii (ATCC 29544), Staphylococcus aureus (DSM 20231), Clostridium perfringens (DSM 756) and Salmonella enterica serovar Typhimurium (ATCC 12028).
The L. rhamnosus isolates were grown in de Man, Rogosa and Sharpe broth (Oxoid Limited, Basingstoke, Hampshire, UK) and incubated at 37°C under anaerobic conditions (10 % H2, 10 % CO2 and 80 % N2; Concept 400 anaerobic chamber, Ruskin Technology, Leeds, UK). For adhesion, competitive exclusion, displacement and inhibition assays, the isolates were grown for 18 h, harvested and then washed twice with PBS buffer. All micro-organisms were metabolically labelled by the addition of 10 μl/ml tritiated thymidine (5-3H-thymidine 1·0 mCi/ml; Amersham Biosciences, Little Chalfont, Bucks, UK) to the media.
Species identity of isolates by partial sequence analysis of the 16S rRNA gene
The L. rhamnosus isolates used in the present study were identified at the species level by partial sequence analysis of the 16S rRNA gene, followed by Blast analysis. In brief, micro-organisms were grown overnight, 1 ml of cells was harvested and the DNA was extracted using the GenElute™ Bacterial Genomic DNA Kit (Sigma, St Louis, MO, USA) following the manufacturer's instructions. Partial amplification of the 16S rRNA gene and species identification were carried out as described previously(Reference Gueimonde, Delgado and Mayo11). Amplified PCR products were purified using the GenElute™ PCR Clean-Up Kit (Sigma), and automated sequencing of the amplicons was carried out at Secugen SL (Madrid, Spain) in an automated sequencer ABI Prism (Applied Biosystems, Foster City, CA, USA). The sequences obtained were compared with those held at the GenBank database by using the Blast application at the NCBI webpage.
Genetic typing of the isolates
Randomly amplified polymorphic DNA-PCR
DNA extracts from the different L. rhamnosus isolates were used for strain typification by randomly amplified polymorphic DNA analysis using previously described conditions and the primer for L. rhamnosus (Reference Tynkkynen, Satokari and Saarela12) (Sigma Genosys, St Louis, MO, USA). PCR assays were run in a Unocycler (VWR International Eurolab S.L., Barcelona, Spain) thermocycler. Amplification products were subjected to electrophoresis on 1 % agarose (Sigma), and the gels were stained and visualised by ethidium bromide staining.
Enterobacterial repetitive intergenic consensus-PCR
Enterobacterial repetitive intergenic consensus-PCR is based on the use of oligonucleotides targeting short repetitive sequences distributed throughout the bacterial genome. DNA was amplified using primers 5′-ATGTAAGCTCCTGGGGATTCAC-3′ and 5′-AAGTAAGTGACTGGGGTGAGCG-3′ (Sigma Genosys) as described previously(Reference Ventura, Meylan and Zink13).
DNA restriction patterns by pulsed-field gel electrophoresis
The L. rhamnosus isolates were typified using analysis of DNA restriction patterns by pulsed-field gel electrophoresis (PFGE). Intact high-molecular weight genomic DNA was isolated and digested in agarose plugs. Cells were grown to an OD600 of 1·5, harvested by centrifugation, washed three times in buffer Tris–HCl (10 mm)–EDTA (1 mm), pH 8·0, and resuspended in 500 μl of the same solution. To form agarose plugs, the cell suspension was heated to 50°C, mixed with an equal volume of 2 % PFGE agarose (Bio-Rad Laboratories, Richmond, CA, USA) in 0·5 × Tris–borate–EDTA buffer and added to moulds. The plugs were incubated for 24 h at 37°C in 1 ml lysis buffer (per plug) containing 50 mm-EDTA (pH 8·0), N-laurylsarcosine (0·5 mg/ml; Sigma), Brij58 (5 mg/ml; Sigma), deoxycholate (2 mg/ml; Sigma), lysozyme (2 mg/ml; Sigma), mutanolysin (15 U/ml; Sigma) and RNase (2 μg/ml; Sigma) and were then deproteinised by incubation at 50°C for 24 h in a solution containing 0·5 m-EDTA pH (8·0), 40 mm-Tris–HCl (pH 8·0), 1 % (w/v) SDS (Sigma) and proteinase K (1·5 mg/ml; Sigma). They were finally washed for 1 h in Tris–HCl (10 mm)–EDTA (1 mm), pH 8·0, and incubated for 24 h at 37°C in Tris–HCl (10 mm)–EDTA (1 mm), pH 8·0, containing Pefabloc SC (0·29 mg/ml; Merck, Darmstadt, Germany).
Thin slices of the agarose plugs were cut and washed six times for 30 min at room temperature in Tris–HCl (10 mm)–EDTA (1 mm), pH 8·0, buffer. DNA within the plugs was digested with 20 U of the appropriate restriction enzyme in 200 μl of the buffer recommended by the supplier. Two different restriction enzymes, SfiI and NotI, were tested. Electrophoresis was carried out at 6 V/cm and 14°C using a CHEF DRII apparatus (Bio-Rad Laboratories) in 1 % PFGE certified agarose (Bio-Rad Laboratories) gels with 0·5 × Tris–borate–EDTA buffer. Pulse times ranged from 2 to 30 s during the 24 h electrophoresis. A DNA pulse marker (LowRange PFG Marker N0350S; New England Biolabs, Hitchin, Herts, UK) was used as molecular size standard. Gels were stained and visualised by ethidium bromide staining.
Characterisation of the isolates by carbohydrate fermentation profiles
The fermentation ability of the L. rhamnosus isolates was obtained in API 50 CH strips (Bio-Mérieux, Marcy l'Etoile, France) following the manufacturer's instructions.
Tolerance to acid
Bacterial cultures (5 ml) were grown overnight, cells were harvested, washed twice with 0·85 % NaCl and resuspended in 500 μl of the same solution; 100 μl of the bacterial suspensions (approximately 1 × 108 cells) were added to 900 μl of simulated gastric juice (125 mm-NaCl, 7 mm-KCl, 45 mm-NaHCO3 and pepsin (3 g/l; Sigma)) adjusted to pH 2·0 or 2·5 with HCl. Suspensions were then incubated for 90 min. Plate counts were made at time point 0 and after 90 min of incubation.
In vitro assay of adhesion to human intestinal mucus
Human intestinal mucus was obtained as described elsewhere(Reference Ouwehand, Salminen and Tolkko3) from the human colon. Colonic mucus was dissolved (0·5 mg protein/ml) in HEPES–Hanks buffer (10 mm-HEPES, pH 7·4). Radiolabelled bacterial absorbance (A 600 nm) was adjusted to 0·25 (sd 0·05) to standardise the bacterial concentration (108 cells/ml). The adhesion assessment of the L. rhamnosus isolates and bacterial pathogens was carried out as described previously(Reference Collado, Meriluoto and Salminen14). Adhesion was expressed as the percentage of radioactivity recovered after adhesion relative to the radioactivity of the bacterial suspension added to the immobilised mucus. Adhesion was determined in three independent experiments, and each assay was performed in triplicate to calculate intra-assay variation.
Exclusion by inhibition assay
To test the ability of the L. rhamnosus isolates to inhibit the adhesion of pathogens, the procedure described by Collado et al. (Reference Collado, Meriluoto and Salminen14, Reference Collado, Grzeskowiak and Salminen15) was used. In brief, unlabelled isolates (108 cells/ml) were added to the wells and incubated for 1 h at 37°C; they were then removed by washing with HEPES–Hanks buffer. Radiolabelled pathogens (108 cells/ml) were then added to the wells and incubated at 37°C for 1 h. Thereafter, the wells were washed, and the bound bacteria were recovered after lysis. Radioactivity was measured by liquid scintillation. The percentage of adhesion inhibition was calculated as the difference between the adhesion of the pathogen in the absence and presence of the different isolates. Inhibition was determined in three independent experiments, and each assay was performed in triplicate.
Exclusion by displacement assay
The ability of the L. rhamnosus isolates studied to displace pathogens already adhered was assessed according to Collado et al. (Reference Collado, Meriluoto and Salminen14, Reference Collado, Grzeskowiak and Salminen15). Radiolabelled pathogens were added to the wells containing mucus. After washing and removal of unbound pathogens, non-radiolabelled isolates were added. Wells were incubated and washed; thereafter, bound bacteria were recovered after lysis and radioactivity was measured. Displacement of pathogens was calculated as the difference between the adhesion of pathogens before and after the addition of the L. rhamnosus isolates. At least three independent experiments were carried out. Each assay was performed in triplicate to calculate intra-assay variation.
Exclusion by competition assay
Competitive inhibition of the model pathogens by the L. rhamnosus isolates studied was determined as described previously(Reference Collado, Grzeskowiak and Salminen15). For the competition test, equal quantities of a given bacterial suspension of isolates and radiolabelled pathogens were mixed and then added to the intestinal mucus and incubated as indicated previously. The cells of the pathogen bound to the mucus were then removed, and adhesion was calculated as described earlier.
Statistical analysis
The adhesion (%) measured in different conditions was the primary endpoint. One-way ANOVA was used to test the overall difference in the adhesion properties between strains. When P < 0·10, Dunnett's (two-tailed) t test was used to compare each strain with the reference strain L. rhamnosus GG without any other pairwise comparisons. The cut-off point was not strictly determined at 0·05 to indicate statistical significance due to the small sample size and large type II error. Based on Dunnett's t test, the exact P values are given, and in which both 0·05 and 0·10 were used as cut-off points to indicate the degree of statistical significance in the difference between strains.
Statistical analysis was conducted using SPSS 18.0 software (SPSS, Inc., Chicago, IL, USA).
Results
Identification at the species level
All L. rhamnosus isolates used were confirmed as members of the species L. rhamnosus by partial sequencing of the 16S rDNA. Sequence analysis showed 100 % sequence homology among the different L. rhamnosus isolates studied as well as with L. rhamnosus 16S rDNA sequences held in the databases (data not shown).
Genotypic and phenotypic characterisation of the isolates
All L. rhamnosus isolates, except the reference strain OL, showed randomly amplified polymorphic DNA, enterobacterial repetitive intergenic consensus and PFGE profiles identical to that of the original L. rhamnosus GG isolate obtained from Professors Gorbach and Goldin. These three techniques allowed differentiation of the isolate L. rhamnosus OL from the other L. rhamnosus isolates.
With regard to phenotypic characterisation, none of the L. rhamnosus isolates fermented glycerol, erythritol, l-arabinose, d-xylose, l-xylose, d-adonitol, methyl-β-d-xylopyranoside, inositol, methyl-α-d-mannopyranoside, d-melibiose, d-saccharose, inulin, d-raffinose, starch, glycogen, xylitol, d-turanose, d-lyxose, d-fucose, d-arabitol, l-arabitol, 2-ketogluconate or 5-ketogluconate. All isolates fermented d-arabinose, d-ribose, d-galactose, d-glucose, d-mannitol, d-fructose, d-mannose, dulcitol, d-sorbitol, N-acetyl glucosamine, amygdalin, arbutin, esculin, salicin, d-cellobiose, d-trehalose, d-melezitose, gentiobiose, d-tagatose, l-fucose and gluconate. All putative L. rhamnosus isolates showed identical sugar fermentation profiles, being different from those of the reference strain OL, which in addition was able to ferment l-sorbose, l-rhamnose, methyl-α-d-glucopyranoside, d-maltose and d-lactose.
Low pH tolerance
Tolerance to acidic pH (pH 2 and 2·5) varied from 43·0 to 79·1 % and showed no significant differences (P>0·05) among the tested L. rhamnosus isolates.
In vitro adhesion assay to colonic mucus
All the tested L. rhamnosus isolates had a good ability to adhere to human colonic mucus, although this varied between different isolates. The adherence of the original L. rhamnosus GG was 19 (sd 7·5) %, and that of the other isolates varied from 12·1 to 24·3 %. Only the adhesion of the reference strain OL differed significantly from that of the original L. rhamnosus GG (P = 0·01), and it also showed the lowest percentage of adhesion among all the isolates tested (data not shown).
Among the pathogenic strains tested, the most marked ability to adhere to colonic mucus was detected for S. aureus (10·2 (sd 3·0) %), followed by C. perfringens (4 (sd 3·1) %), S. enterica serovar Typhimurium (2·4 (sd 0·7) %) and C. sakazakii (2 (sd 0·7) %).
Exclusion by inhibition assay
The ability to inhibit the adhesion of pathogens to colonic mucus by the L. rhamnosus isolates differed significantly (see Fig. 1). The original L. rhamnosus GG was able to inhibit all pathogens tested, and this micro-organism reduced the adhesion of C. sakazakii (31 %), S. enterica serovar Typhimurium (25 %), C. perfringens (25 %) and S. aureus (24 %).
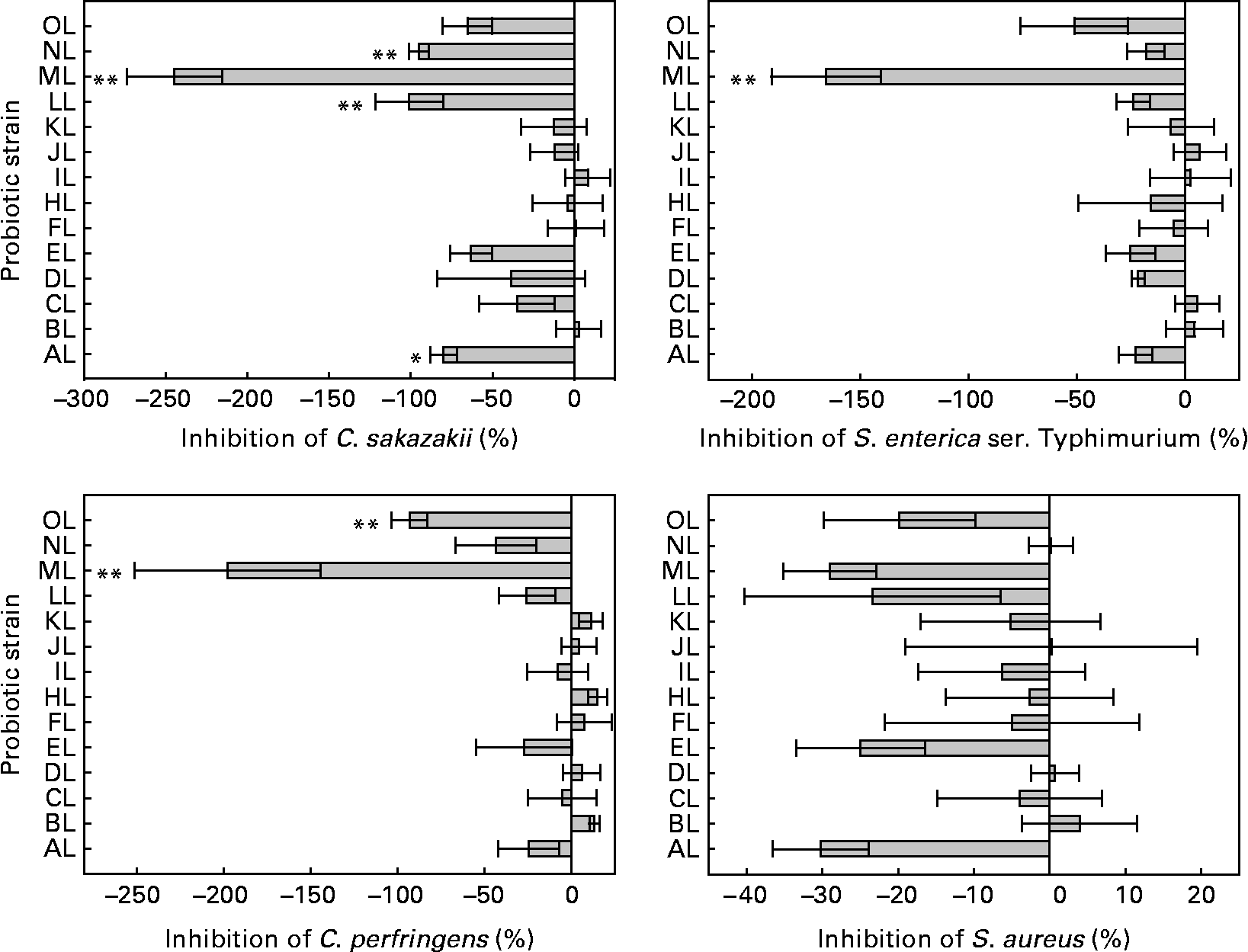
Fig. 1 Inhibition of the adhesion to colonic mucus of pathogenic bacteria by the tested Lactobacillus rhamnosus isolates. Results are expressed as percentages with regard to the inhibition obtained with the original L. rhamnosus GG isolate donated by Professor S. Gorbach. Values on the X-axis higher than 0 indicate adhesion inhibition higher than the original L. rhamnosus GG, and those lower than 0 indicate adhesion inhibition lower than the original L. rhamnosus GG. Adhesion inhibition significantly different from the original L. rhamnosus GG (* if 0·05 ≤ P < 0·10 and ** if P < 0·05). Values are means, with standard errors represented by vertical bars. The OL indicates the external reference L. rhamnosus strain. C. sakazakii, Cronobacter sakazakii; S. enterica ser., Salmonella enterica serovar; C. perfringens, Clostridium perfringens; S. aureus, Staphylococcus aureus. Identification codes: AL, BL, CL, DL, EL, FL, HL, IL, JL, KL, LL, ML and NL.
Differences were between L. rhamnosus isolates in the adhesion of C. sakazakii, S. enterica serovar Typhimurium and C. perfringens (ANOVA, P < 0·001 for each) but not in the adhesion of S. aureus (P = 0·352).
Four L. rhamnosus isolates (AL, LL, ML and NL) showed different inhibition (P = 0·083, P = 0·015, P < 0·001, P = 0·025, respectively) of C. sakazakii compared with the original L. rhamnosus GG. The isolate ML, again, differed (P < 0·001) from the original L. rhamnosus GG in the adhesion inhibition of S. enterica serovar Typhimurium. Two L. rhamnosus isolates (ML and OL) differed (P < 0·001, P = 0·032, respectively) in the adhesion inhibition of C. perfringens from the original L. rhamnosus GG. The adhesion inhibition of S. aureus did not differ between the tested L. rhamnosus isolates and the original L. rhamnosus GG.
Exclusion by displacement assay
All L. rhamnosus isolates were able to displace the preadhered model pathogens tested in colonic mucus. Differences were between L. rhamnosus isolates in the adhesion of C. sakazakii, S. enterica serovar Typhimurium (ANOVA, P = 0·063, 0·001, respectively) but not between C. perfringens and S. aureus (ANOVA, P = 0·128, 0·717, respectively). The original L. rhamnosus GG displaced 54 % of adhered S. enterica serovar Typhimurium, and it differed (P = 0·084) from the L. rhamnosus LL isolate. C. perfringens was displaced by 33 % and S. aureus by 20 % by the original L. rhamnosus GG, and all the other isolates showed no difference in the displacement from the original L. rhamnosus GG. The original L. rhamnosus GG displaced C. sakazakii by 12 %, differing (P = 0·082, 0·009, 0·017, 0·013, 0·068, 0·019, 0·01, 0·092, 0·051, respectively) from the other study isolates AL, BL, CL, DL, EL, HL, IL, KL and NL. The results are shown in Fig. 2.
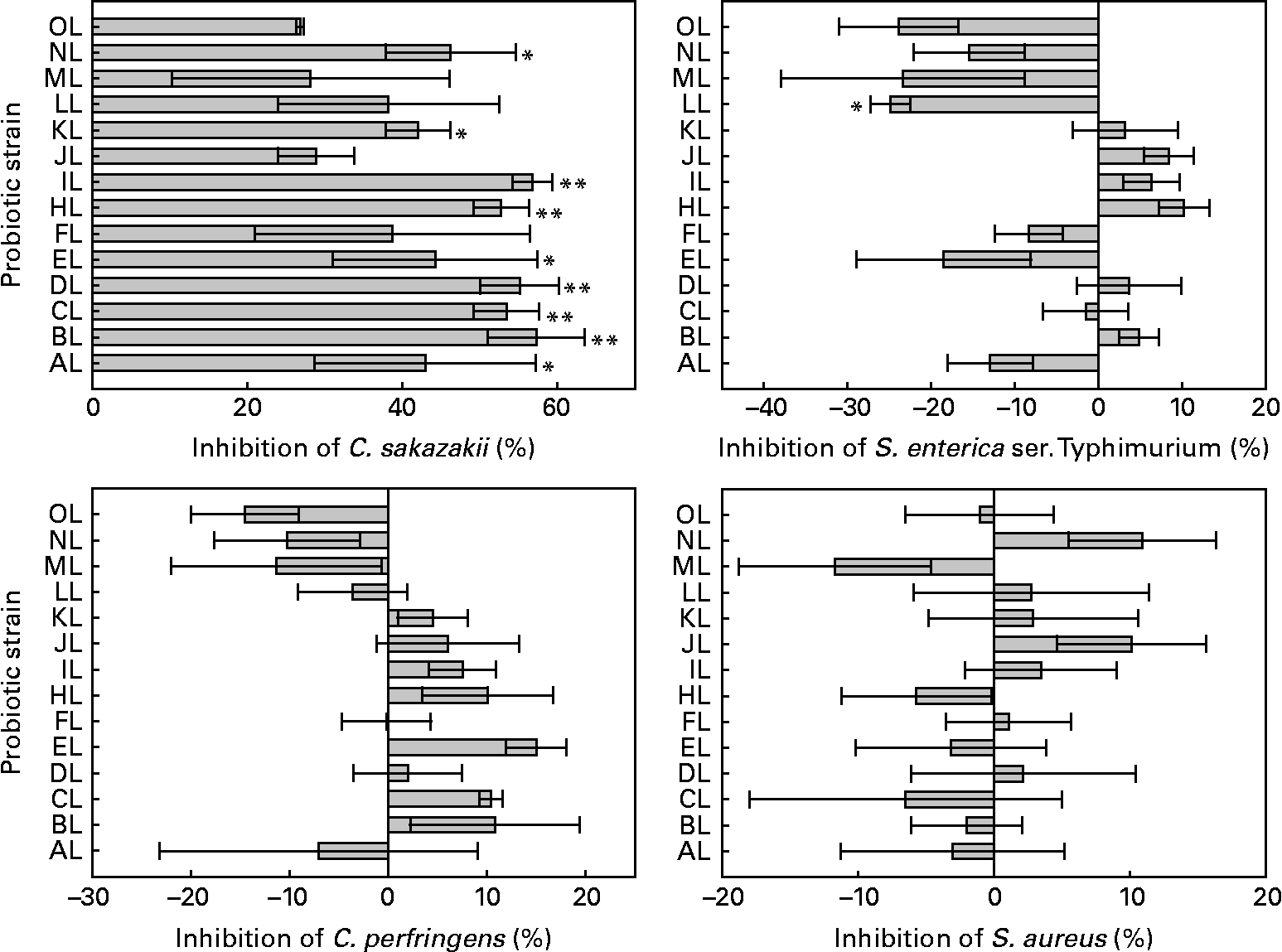
Fig. 2 Displacement of the adhesion to colonic mucus of pathogenic bacteria by the tested Lactobacillus rhamnosus isolates. Results are expressed as percentages with regard to the displacement obtained with the original L. rhamnosus GG isolate donated by Professor S. Gorbach. Values on the X-axis higher than 0 indicate adhesion displacement higher than the original L. rhamnosus GG, and those lower than 0 indicate adhesion displacement lower than the original L. rhamnosus GG. Adhesion displacement significantly different from the original L. rhamnosus GG (* if 0·05 ≤ P < 0·10 and ** if P < 0·05). Values are means, with standard errors represented by vertical bars. The OL indicates the external reference L. rhamnosus strain. C. sakazakii, Cronobacter sakazakii; S. enterica ser., Salmonella enterica serovar; C. perfringens, Clostridium perfringens; S. aureus, Staphylococcus aureus. Identification codes: AL, BL, CL, DL, EL, FL, HL, IL, JL, KL, LL, ML and NL.
Exclusion by competition assay
The inhibition by competition varied among the L. rhamnosus isolates and pathogens tested. The relevant data are shown in Fig. 3. The presence of some isolates increased the adhesion of C. sakazakii, S. enterica serovar Typhimurium and C. perfringens to colonic mucus. All but one isolate (BL) was able to reduce the adhesion of S. aureus. Differences were between L. rhamnosus isolates in the adhesion of C. sakazakii, S. enterica serovar Typhimurium, C. perfringens and S. aureus (ANOVA, P < 0·001, P < 0·001, P = 0·005, P = 0·023, respectively). The original L. rhamnosus GG showed inhibition abilities for all pathogens tested. This micro-organism inhibited the adhesion of C. perfringens in 47 %, being different (P = 0·057, 0·059, 0·07, 0·091, respectively) from the BL, LL, ML and NL isolates; S. aureus in 39 %, being different (P = 0·019) from the BL isolate; S. enterica serovar Typhimurium in 5 %, being different (P = 0·064, 0·014, 0·026, respectively) from the IL, ML and NL isolates; C. sakazakii in 9 %, showing a difference (P = 0·001, P = 0·003, P = 0·048, P = 0·001, P < 0·001, P = 0·079, respectively) from the AL, DL, LL, ML, NL isolates and the reference strain OL.
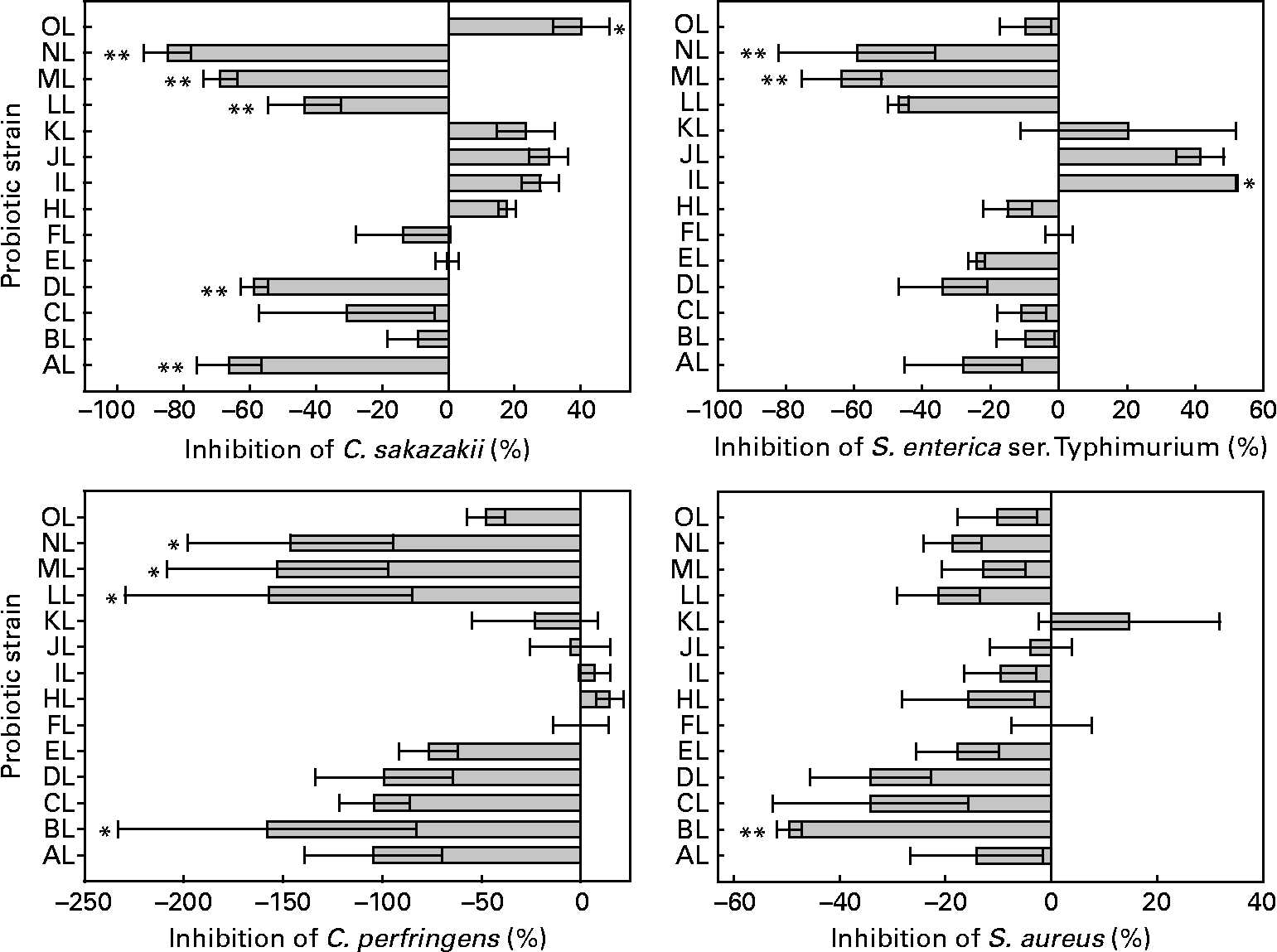
Fig. 3 Competition of the adhesion to colonic mucus of pathogenic bacteria by the tested Lactobacillus rhamnosus isolates. Results are expressed as percentages with regard to the competition obtained with the original L. rhamnosus GG isolate donated by Professor S. Gorbach. Values on the X-axis higher than 0 indicate adhesion competition higher than the original L. rhamnosus GG, and those lower than 0 indicate adhesion competition lower than the original L. rhamnosus GG. Adhesion competition significantly different from the original L. rhamnosus GG (* if 0·05 ≤ P < 0·10 and ** if P < 0·05). Values are means, with standard errors represented by vertical bars. The OL indicates the external reference L. rhamnosus strain. C. sakazakii, Cronobacter sakazakii; S. enterica ser., Salmonella enterica serovar; C. perfringens, Clostridium perfringens; S. aureus, Staphylococcus aureus. Identification codes: AL, BL, CL, DL, EL, FL, HL, IL, JL, KL, LL, ML and NL.
Discussion
The term ‘probiotic’ per se implies a health benefit. Based on the definition of probiotics, consumption of these bacteria present in food products should exert a range of health-promoting effects in the host(1). In several meta-analyses and reviews, however, strains with no such documentation are reported along with true probiotics(Reference Taylor, Dunstan and Prescott16, Reference D'Souza, Rajkumar and Cooke17). Moreover, confusion is caused by reporting on the effect of probiotic strains and their combinations together. Indeed, scientifically it is not surprising that different micro-organisms have different effects.
We demonstrate in the present study, for the first time, the differences in the in vitro properties of several L. rhamnosus GG isolates from different probiotic products. As a preceding step, all micro-organisms were genotypically and phenotypically characterised to ensure that all putative L. rhamnosus GG isolates had identical profiles, obviating any contamination or misidentification of the isolates used in the different products. All putative L. rhamnosus GG isolates were members of the L. rhamnosus group proved by the use of profiling techniques such as randomly amplified polymorphic DNA-PCR, enterobacterial repetitive intergenic consensus-PCR and PFGE, and they showed profiles identical to that of the original L. rhamnosus GG isolate. The genotypic and phenotypic characterisation allowed distinction of the external reference strain OL from the L. rhamnosus isolates.
Adherence of bacteria to intestinal mucus or epithelium is known to be a prerequisite for colonisation and infection of the gastrointestinal tract by many pathogens(Reference Finlay and Falkow18). The adherent probiotic strains may at least temporarily colonise the gastrointestinal tract and inhibit or compete with pathogens(Reference Vesterlund, Karp and Salminen19). Several reports have described the adhesion abilities of probiotic strains and also their inhibitory effects on model pathogen adhesion to human mucus(Reference Collado, Meriluoto and Salminen14, Reference Ouwehand, Tuomola and Tolkko20, Reference Collado, Meriluoto and Salminen21). Variations in these properties may affect the clinical outcome of intervention studies. In the present study, all tested L. rhamnosus isolates showed the ability to adhere to human colonic mucus, and the adhesion ability of different isolates varied but did not differ significantly. Potentially, the preculturing may have decreased the differences, and thus other adhesion assays without the preculturing step should be considered. However, all isolates had significantly higher adhesion ability than that of the reference strain.
The model pathogen exclusion results varied significantly and depended on the isolate in question. The results indicate differences in these important properties of probiotics among the different L. rhamnosus isolates obtained from different sources. Moreover, conducting our analyses required growing the different isolates under the same standardised laboratory conditions. Despite this culture step, which may have decreased the differences in physiological properties of the strain or the effect of different excipients used during manufacturing of the products, our observations suggest that the in vitro properties of probiotics may vary depending on the production conditions used for the same probiotic strain.
The present findings indicate that different isolates of the same strain may possess different properties which may influence their in vivo effects, underlining the importance of control of the manufacturing process and the food matrix. The L. rhamnosus isolates tested in the present study were chosen to be from different products and origins. The manufacturing process may thus have had a significant impact on the strain properties. Often probiotics are used in fermented dairy products or as freeze-dried preparations. Both provide a matrix for the micro-organisms, and previous studies have indicated that the matrix may affect the strain properties(Reference Kankaanpaa, Salminen and Isolauri22, Reference Kankaanpaa, Yang and Kallio23). This, along with the production conditions, may in part explain the different results obtained between strain isolates when characterising strain impact on pathogen adhesion to mucus, although the existence of point mutations in some of the different isolates cannot be overruled.
In addition to the differences between strains, Deepika et al. (Reference Deepika, Green and Frazier24) have reported that the adhesion of the L. rhamnosus strain GG to Caco-2 cells was the highest when bacteria collected during the early stationary phase of growth were used. This finding suggests that the early stationary phase may be the optimum harvest point for Lactobacillus GG to obtain the most adhesive strain.
Though L. rhamnosus GG is one of the probiotics subjected to the largest number of human intervention studies, in many cases no attention has been paid to the specific properties of the strains used. The lack of such reports is surprising, especially considering the early findings on changes in the in vitro properties of early L. rhamnosus GG strains following different production processes and a long-term series of reinoculations, suggesting deterioration of adhesion properties(Reference Elo, Saxelin and Salminen6) and the report on production lot differences in an L. acidophilus strain influencing the outcome of human intervention studies(Reference Clements, Levine and Ristaino8).
Taken together, we conclude that the original properties used in the selection of specific probiotic strains may indeed be influenced by industrial production processes and conditions as well as by the food matrix used. This finding sets important prerequisites for quality control in probiotics. Ensuring the original properties is especially important when the strain or product is used in human intervention studies, as small changes may significantly influence the outcome. The finding also presupposes new quality-control measures for the manufacture of probiotics for food use to preserve the original properties, which may have an impact on efficacy in human studies.
Acknowledgements
The present study was supported by the Academy of Finland (grant no. 126835) and a grant from Mead Johnson Nutrition. None of the authors had a personal or financial conflict of interest. The author's responsibilities were as follows: Ł. G., E. I., S. S. and M. G. planned and coordinated the study; Ł. G. and M. G. were responsible for the laboratory experiments of the study. All authors participated in the analysis of results and in writing the manuscript. We thank Tuija Poussa for statistical consultation during the data analysis and help in writing the manuscript.



