


The human intestinal microbiota plays a key role in numerous metabolic, physiological, nutritional and immunological processes( Reference O'Hara and Shanahan 1 ), and perturbations in the composition of the microbiota influences human health( Reference O'Toole and Claesson 2 ). Much of the early information regarding the intestinal microbiota has come from studies that used culture-dependent techniques, which reveal only a minority of species constituting the microbial population( Reference O'Toole and Claesson 2 , Reference Adlerberth and Wold 3 ). However, the advent of culture-independent, DNA-based analyses has generated data that can be mined for information on the composition and functional properties of this hitherto-uncultured microbiota( Reference O'Toole and Claesson 2 , Reference Rastall 4 , Reference Dusko Ehrlich 5 ).
The microbial content of the gastrointestinal tract (GIT) changes along its length, ranging from a narrow diversity and low numbers of microbes in the stomach to a wide diversity and high numbers in the large intestine( Reference Tiihonen, Ouwehand and Rautonen 6 , Reference Isolauri, Salminen and Ouwehand 7 ) (Fig. 1). The best-studied region of the gut is the distal colon, and in adults, faeces-derived populations have been estimated to consist of 1013 to 1014 micro-organisms, composed of approximately 1100 prevalent species, with at least 160 such species per individual. In its entirety, the microbiota is estimated to contain 150-fold more genes than the human genome( Reference Qin, Li and Raes 8 ). The majority of bacteria belong either to the phylum Firmicutes (including Clostridium, Enterococcus, Lactobacillus and Ruminococcus) or to the phylum Bacteroidetes (including Bacteroides and Prevotella genera), which constitute over 90 % of the known phylogenetic categories found in the human intestine( Reference Qin, Li and Raes 8 – Reference Rajilić-Stojanović, Heilig and Molenaar 14 ). Although there is huge inter-individual variability in microbial compositions( Reference Qin, Li and Raes 8 , Reference Eckburg, Bik and Bernstein 9 , Reference Hayashi, Sakamoto and Benno 12 , Reference Hayashi, Sakamoto and Kitahara 15 ), recent work has revealed that a core group of more than fifty taxa can be found in nearly half of the human subjects sampled( Reference Qin, Li and Raes 8 , Reference Tap, Mondot and Levenez 13 ). It has also been suggested that the microbiota of most individuals can be categorised into three predominant variants, or ‘enterotypes’, dominated by three different genera: Bacteroides; Prevotella; Ruminococcus, which are independent of age, sex, nationality and BMI( Reference Arumugam, Raes and Pelletier 16 ). This concept was partially supported by Wu et al. ( Reference Wu, Chen and Hoffmann 17 ), who identified two enterotypes, distinguished primarily by the levels of Bacteroides and Prevotella, which were largely driven by diet. More recently, considerable debate has arisen about the notion of enterotypes( 18 , Reference Jeffery, Claesson and O'Toole 19 ), with a number of studies( Reference Claesson, Jeffery and Conde 20 , Reference Huse, Ye and Zhou 21 ) failing to identify the three distinct categories described by Arumugam et al. ( Reference Arumugam, Raes and Pelletier 16 ). Researchers are now favouring the idea of a continuum or gradient of species functionality rather than a discontinuous variation with segregated types( Reference Jeffery, Claesson and O'Toole 19 ).
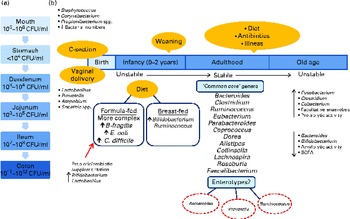
Fig. 1 (a) Variations in microbial numbers across the length of the gastrointestinal tract. (b) Selected features affecting the establishment and maintenance of the microbiota and factors influencing the composition of the microbiota. Micro-organisms are listed where their abundance is related to a particular environmental factor( Reference Tiihonen, Ouwehand and Rautonen 6 – Reference Qin, Li and Raes 8 , Reference Tap, Mondot and Levenez 13 , Reference Arumugam, Raes and Pelletier 16 , Reference Marques, Wall and Ross 25 , Reference Morelli 26 , Reference Cusack and O'Toole 45 , Reference Dominguez-Bello, Costello and Contreras 173 ). C-section, Caesarean section; CFU, colony-forming units; B. fragilis, Bacteroides fragilis; E. coli, Escherichia coli; C. difficile, Clostridium difficile.
Studies have also identified a core microbiome at the gene rather than at the organismal lineage level( 22 , Reference Turnbaugh, Hamady and Yatsunenko 23 ). These studies suggest that, rather than a core group of species, individuals share a core group of microbiome functions and individuals exhibiting particular phenotypes (e.g. obese or non-obese) may display different patterns of gut microbes but share a core group of functions( Reference Turnbaugh, Hamady and Yatsunenko 23 ). Changes in this core set of genes may account for different states of health and disease. Future research will investigate whether the metagenome predicts a risk for developing particular human diseases to obtain new microbial diagnostic markers that may allow early diagnosis of diseases and development of potentially new therapeutic strategies.
Although much has been discovered in the last decade about the intestinal microbiota, there are biases and limitations to the current knowledge related to study design, sample collection and confounding variables, such as diet. Investigations into the impact of diet on the intestinal microbiota are challenged by the inability of researchers to carry out large-scale, carefully controlled trials in humans. There is also a need to better clarify the mechanisms through which changes in microbial dysbiosis promote disease states to improve our understanding of the causal relationship between the gut microbiota and disease. Whether a disease-prone microbial composition can be transformed into a healthier composition by probiotic/prebiotic/dietary interventions remains a fundamental unanswered question. Nonetheless, the emergence and growing accessibility of the next-generation sequencing technologies will greatly advance the discovery of the composition and functional capacity of microbial communities in the human gut. In the present review, we discuss recent insights into the impact of age, diet, antibiotic use and disease on the intestinal microbiota. We also highlight the role of probiotics, in particular, the potential role of dead or inactivated microbes as a therapeutic modality in the treatment and prevention of diseases and/or restoration of human health.
Factors influencing the composition of the microbiota
Age
The development of the human microbiota is a dynamic process, with individuals exhibiting differences in terms of microbial diversity and variation at different life stages (Fig. 1)( Reference O'Toole and Claesson 2 , Reference Tiihonen, Ouwehand and Rautonen 6 , Reference Biagi, Candela and Fairweather-Tait 24 ). The human microbiota is established at birth when the intestine becomes inhabited by a population that is characterised by instability( Reference Tiihonen, Ouwehand and Rautonen 6 ). Initially, facultative anaerobes such as Enterobacteriaceae, streptococci and staphylococci dominate( Reference Marques, Wall and Ross 25 ). In recent years, more stringent hygienic conditions during delivery combined with shorter hospital stays have reduced bacterial exposure, leading to changes in the initial colonisation pattern, with skin-derived staphylococci being the first colonisers of the infant gut rather than faecal Enterobacteriaceae( Reference Marques, Wall and Ross 25 , Reference Morelli 26 ). For infants born vaginally, the first encounter with micro-organisms occurs in the birth canal, where colonisation is initiated by the maternal vaginal and intestinal microbiota as well as the environment( Reference Lupp and Finlay 27 ). In contrast, for infants delivered by caesarean section, the environment (e.g. nursing staff and the air) is an extremely important source of colonising bacteria, and these infants have lower intestinal bacterial counts with less diversity in the early weeks of life( Reference Morelli 26 , Reference Grölund, Lehtonen and Eerola 28 , Reference Axad, Konya and Maughan 29 ).
Other factors influencing the microbiota include gestational age, hospitalisation of the infant, antibiotic use and infant feeding. Breast-fed infants have a microbiota dominated by Bifidobacterium ( Reference Bezirtzoglou, Tsiotsias and Welling 30 – Reference Yatsunenko, Rey and Manary 33 ) and Ruminococcus ( Reference Morelli 26 , Reference Favier, Vaughan and De Vos 34 ), with the rates of colonisation by Escherichia coli, Clostridium difficile, Bacteroides fragilis group bacteria and lactobacilli being significantly lower than those observed in exclusively formula-fed infants( Reference Penders, Thijs and Vink 31 , Reference Yoshioka, Iseki and Fujita 35 ). The microbiota of formula-fed infants is more complex( Reference Bezirtzoglou, Tsiotsias and Welling 30 , Reference Agans, Rigsbee and Kenche 36 ) and comprises a variety of bacterial genera, including enterobacterial genera, Streptococcus, Bacteroides and Clostridium, as well as Bifidobacterium ( Reference Favier, Vaughan and De Vos 34 ) and Atopobium ( Reference Bezirtzoglou, Tsiotsias and Welling 30 ). It must be noted that some reports have found no differences in the compositions of the microbiota between breast-fed and formula-fed infants and have attributed this to modern formulas more closely mimicking the composition of human breast milk( Reference Adlerberth and Wold 3 ). The composition of the microbiota changes further with the introduction of solid foods, and a complex, more stable community similar to the adult microbiota becomes established after weaning (at 2–3 years of age)( Reference Yatsunenko, Rey and Manary 33 , Reference Favier, Vaughan and De Vos 34 , Reference Collins and Gibson 37 , Reference Koenig, Spor and Scalfone 38 ).
Throughout adulthood, the composition of the intestinal microbiota is relatively stable and is only transiently altered by external disturbances( Reference Delgado, Suárez and Mayo 39 ), as will be discussed below. However, this relative stability is reduced in old age( Reference McCartney, Wenzhi and Tannock 40 ). There is considerable variation in the reported microbial compositions of elderly subjects, which appear to be dependent on residence cohort, geographical location and detection methods used( Reference Tiihonen, Ouwehand and Rautonen 6 , Reference Mueller, Saunier and Hanisch 41 ). The large inter-individual variation in microbial compositions continues into old age( Reference Claesson, Cusack and O'Sullivan 42 ), and the process of ageing coincides with the decreasing diversity of the microbiota( Reference Biagi, Nylund and Candela 43 ). Researchers are continually striving to elucidate the composition of the intestinal microbiota of the elderly, but as yet no specific ‘common core’ has been identified. However, some of the fundamental changes that occur include a decrease in the total number and species diversity of bifidobacteria and Bacteroides as well as a reduction in amylolytic activity and the availability of total SCFA. There is a concurrent increase in the number of facultative anaerobes, fusobacteria, clostridia and eubacteria as well as an increase in proteolytic activity( Reference Woodmansey 44 , Reference Cusack and O'Toole 45 ). A study by the ELDERMET consortium has found that the microbial population of elderly Irish subjects is dominated by Bacteroidetes, whereas the microbiota of younger subjects is dominated by Firmicutes( Reference Claesson, Cusack and O'Sullivan 42 ). However, Biagi et al. ( Reference Biagi, Nylund and Candela 43 ) did not find significant differences among the Firmicutes:Bacteroidetes ratios of Italian centenarians, elderly and young adults. These conflicting results have been attributed to the country-related variation in the compositions of the gut microbiota( Reference Biagi, Candela and Fairweather-Tait 24 ), which has been highlighted several years ago( Reference Mueller, Saunier and Hanisch 41 ) and presumably may be linked to the diet. Furthermore, the composition of the gut microbiota of the elderly may also vary depending on residence location( Reference Claesson, Jeffery and Conde 20 , Reference Bartosch, Fite and Macfarlane 46 ), which is a proxy measure for radically different diets (see below).
Diet
Diet is a factor that undoubtedly influences the composition of the intestinal microbiota. Diet provides nutrients for both the host and the bacteria in the GIT. Most of the enzymes needed to break down the structural polysaccharides in plant material are not encoded by mammalian genomes. The intestinal microbiota produces a larger collection of degradative enzymes and exhibits a broader range of metabolic capabilities( Reference Flint, Scott and Duncan 47 ). It is estimated that 20–60 g of dietary carbohydrates reach the colon on a daily basis( Reference Flint, Scott and Duncan 47 ), including resistant starch, NSP, plant cell wall polysaccharides and non-digestible oligosaccharides( Reference Flint, Scott and Duncan 47 – Reference Scott, Duncan and Flint 49 ). Some dietary proteins (e.g. collagen and elastin) as well as various secondary plant metabolites (e.g. polyphenolic substances) can also reach the large intestine and may undergo bacterial transformations( Reference Louis, Scott and Duncan 50 , Reference Guarner and Malagelada 51 ).
Alternative substrates can give rise to different products due to fermentation via different metabolic processes, while the same substrate can be metabolised by different pathways depending on the rate of supply or the physiology and environment of the bacterial cell( Reference Louis, Scott and Duncan 50 ). Changes in the composition of the gut microbiota in response to dietary intake occur because different bacterial species are better equipped (genetically) to utilise different substrates( Reference Scott, Duncan and Flint 49 ). Generally, bacteria favour carbohydrates as primary energy sources if they are available( Reference Apajalahti 52 ). Metagenomic sequencing of the intestinal microbiota has identified a large group of carbohydrate-active enzymes( Reference Kurokawa, Itoh and Kuwahara 53 ). While certain species, particularly those of the phylum Bacteroidetes, possess large numbers of genes encoding carbohydrate-active enzymes, which allows them to switch between different energy sources, other groups encode fewer carbohydrate-active enzymes and are noticeably more specialised( Reference Flint, Scott and Duncan 47 ). Dietary supplementation with prebiotics such as inulin and fructo-oligosaccharides can promote the growth of specific groups of bacteria, including bifidobacteria( Reference Flint, Duncan and Scott 54 – Reference Roberfroid 56 ). A recent study has demonstrated rapid and reversible changes in the relative abundance of specific dominant bacterial groups after changes in the major type of non-digestible carbohydrate (i.e. resistant starch, NSP or reduced-carbohydrate diet). There were profound inter-individual differences in the response of the microbial community to dietary change due to inter-individual differences in the initial microbial composition; this suggests that dietary advice on the consumption of non-digestible carbohydrates may need to be personalised in the future( Reference Walker, Ince and Duncan 57 ).
Saccharolytic bacterial fermentation mainly takes place in the proximal colon (due to greater availability of fermentable carbohydrates)( Reference Hamer, Jonkers and Venema 58 ) and may result in the production of SCFA( Reference Guarner and Malagelada 51 ), the type and levels of which depend on the source and quantity of carbohydrates available and the microbiota present( Reference Scott, Duncan and Flint 49 ). SCFA are energy sources for the colonic epithelium, and butyrate, in particular, exerts important effects on cell differentiation and gut health( Reference Hamer, Jonkers and Venema 58 ). Proteolytic fermentation generally takes place in the distal colon (where fermentable carbohydrates become depleted)( Reference Hamer, Jonkers and Venema 58 ) and results in the production of SCFA in addition to ammonia, amines, phenols, thiols and indoles( Reference Guarner and Malagelada 51 ).
Early studies comparing dietary patterns (e.g. ‘Japanese’ v. ‘Western’) or examining the impact of changing the proportions of food categories on the intestinal microbiota have found only moderate effects involving a few genera( Reference Drasar, Crowther and Goddard 59 – Reference Drasar, Jenkins and Cummings 61 ). These studies relied on culture-based techniques and were, therefore, limited in their ability to detect changes in the fine detail of the composition of the gut microbiota. More recent studies have employed culture-independent approaches and have further elucidated the role of diet in the determination of the composition of the intestinal microbiota in humans (Table 1).
Table 1 Associations of the human intestinal microbiota with habitual dietary patterns or interventions

↑ , Increased; ↓ , decreased; FISH, fluorescent in situ hybridisation; qPCR, quantitative real-time PCR; DGGE, denaturing gradient gel electrophoresis; KEGG, Kyoto Encyclopedia of Genes and Genomes; OTU, operational taxonomic unit; ↔ , no change.
In a landmark study, De Filippo et al. ( Reference De Filippo, Cavalieri and Di Paola 62 ) compared the faecal microbiota of European children (consuming a ‘Western’ diet) with that of children in the African state of Burkina Faso (consuming a plant-rich, ‘rural’ diet, high in fibre content). The Burkina Faso children had a lower abundance of bacteria of the phylum Firmicutes and a higher abundance of those of the phylum Bacteroidetes (mainly Prevotella and Xylanibacter) in their faecal microbiota compared with the European children, who had higher levels of Enterobacteriaceae. Prevotella and Xylanibacter, which contain genes for cellulose and xylan hydrolysis, were associated with increased levels of faecal SCFA. The authors postulated that the gut microbiota co-evolved with the plant-rich diet of the Burkina Faso children, allowing them to maximise energy extraction from dietary fibre while also protecting them from inflammation and non-infectious intestinal diseases.
Similar dietary associations have been found in a study linking the dietary patterns of American adults with gut microbial enterotypes, dominated by Bacteroides or Prevotella. Wu et al. ( Reference Wu, Chen and Hoffmann 17 ) found that the Bacteroides enterotype was positively associated with protein and animal fat, whereas the Prevotella enterotype was associated with a diet high in carbohydrates and low in meat and dairy products.
Vegetarianism has also been shown to alter the composition of the intestinal microbiota( Reference Hayashi, Sakamoto and Benno 63 – Reference Kabeerdoss, Devi and Mary 65 ). The higher intakes of carbohydrate and fibre associated with this dietary practice result in the production of SCFA by microbes, which lowers the intestinal pH, preventing the growth of potentially pathogenic bacteria such as E. coli and other members of Enterobacteriaceae spp.( Reference Zimmer, Lange and Frick 66 ). Indeed, Zimmer et al. ( Reference Zimmer, Lange and Frick 66 ) demonstrated that subjects consuming a vegan or vegetarian diet had lower stool pH than controls and that total counts of culturable Bacteroides spp., Bifidobacterium spp., E. coli and Enterobacteriaceae spp. were significantly lower in vegan samples than in the controls. A vegetarian diet has also been shown to decrease the amount and change the diversity of Clostridium cluster IV and Clostridium rRNA clusters XIVa and XVII( Reference Hayashi, Sakamoto and Benno 63 , Reference Liszt, Zwielehner and Handschur 64 ).
It has recently been reported that diverse dietary patterns are responsible for the variation in the compositions of the gut microbiota observed between community-dwelling elderly subjects and subjects in long-term residential care. The diet of community-dwelling individuals was typically more diverse with low-to-moderate fat and high fibre intakes, whereas that of subjects in long-term residential care was less diverse with moderate-to-high fat and low-to-moderate fibre intakes. Those in long-term care had a less diverse microbiota with a higher proportion of bacteria of the phylum Bacteroidetes, while community-dwelling subjects had a more diverse microbiota with a higher proportion of bacteria of the phylum Firmicutes. Community-dwelling subjects had a higher abundance of bacteria of the genus Prevotella, supporting the association between Prevotella and a diet high in carbohydrates as observed in the Burkina Faso children and American adults. Coprococcus and Roseburia were also more abundant in the faecal microbiota of community-dwelling subjects, whereas Parabacteroides, Eubacterium, Anaerotruncus, Lactonifactor and Coprobacillus were more abundant in subjects in long-term care. For subjects in long-term care, both the faecal microbiota and diet were associated with the duration of stay, with subjects residing for more than 1 year having diet and microbiota that were furthest separated from those of community-dwelling subjects compared with recently admitted subjects. Interestingly, the major trends in the microbiota that separated the community-dwelling elderly from the elderly in long-term care were associated with changes in frailty, inflammation and other clinical markers and hence indicate a role for diet-driven microbial composition alterations in health among the elderly( Reference Claesson, Jeffery and Conde 20 ).
Changes in the abundance of the gut microbiota of (humanised germ-free (GF)) mice have been analysed after the mice were switched from a diet low in fat and rich in plant polysaccharides to a ‘Western’ diet high in fat and sugar and low in plant polysaccharides. After just a single day, mice on the ‘Western’ diet displayed an increased abundance of bacteria of the phylum Firmicutes and a decreased abundance of those of the phylum Bacteroidetes( Reference Turnbaugh, Ridaura and Faith 67 ). Hildebrandt et al. ( Reference Hildebrandt, Hoffmann and Sherrill-Mix 68 ) also found distinctive changes in the abundance of the gut microbiota of mice following a switch from a standard chow to a high-fat diet, which was associated with a proportional decrease in the abundance of bacteria of the phylum Bacteroidetes and an increase in that of both Firmicutes and Proteobacteria.
Faith et al. ( Reference Faith, McNulty and Rey 69 ) developed a statistical model for predicting how a change in diet would alter the abundance of particular species of the gut microbiota. A model community of ten genome-sequenced human intestinal bacteria (including Bacteroides thetaiotaomicron, Bacteroides ovatus, Bacteroides caccae, E. coli, Desulfovibrio piger, Collinsella aerofaciens, Clostridium symbiosum, Blautia hydrogenotrophica, Eubacterium rectale and Marvinbryantia formatexigens) was introduced into GF mice, and the composition of the intestinal community as the mice consumed different proportions of protein (casein), fat (‘corn’ oil), polysaccharide (‘cornstarch’) and sugar (sucrose) was monitored. Each mouse was fed a randomly selected diet with diet switches occurring every 2 weeks. Steady-state levels of community members were achieved within 4 d of a diet change. Notably, the total DNA yield per faecal pellet increased as the amount of casein in the host diet was increased. In addition, changes in species abundance as a function of changes in casein concentration in the host diet were apparent for all the ten species; the abundance of seven species was positively correlated with casein concentration, whereas that of the remaining three species (E. rectale, M. formatexigens and D. piger) was negatively correlated with casein concentration. Indeed, inspection of the most highly expressed genes of E. rectale and M. formatexigens indicated that they focused on carbohydrate catabolism, whereas D. piger can use only a restricted number of substrates (e.g. lactate, H2 and succinate). In a follow-up experiment involving diets containing various mixtures of puréed human baby foods (i.e. foods more typically consumed in human diets), changes in species abundance that were a function of diet ingredients (e.g. apple, beef, chicken, oat, pea, peach, rice and sweet potato) were found. For example, B. ovatus increased in absolute abundance with an increased concentration of oats in the diet, whereas most of the bacterial species responded to multiple ingredients.
Although these results from animal studies are interesting, it can be difficult to apply these findings to humans due to the artificial nature of these experiments compared with natural human microbiota and food consumption patterns. Only a limited number of human clinical trials have assessed the effects of dietary pattern changes on the intestinal microbiota( Reference Wu, Chen and Hoffmann 17 , Reference Walker, Ince and Duncan 57 , Reference De Palma, Nadal and Collado 70 , Reference Muegge, Kuczynski and Knights 71 ). In a controlled-feeding study with ten individuals (Bacteroides enterotype), Wu et al. ( Reference Wu, Chen and Hoffmann 17 ) found that the composition of the microbiome changed detectably within 24 h of consuming a high-fat/low-fibre or low-fat/high-fibre diet, showing the rapid effect that diet can have on the intestinal microbiota. However, enterotype identity remained constant, with no stable changes in the composition of the Prevotella enterotype, indicating that alternative enterotype states are associated with long-term diet intake.
In overweight men, supplementation of the diet with resistant starch increased the faecal levels of E. rectale and Ruminococcus bromii, which correlated with the fermentation of fibre. However, the inter-individual variation in the responses of the microbiota to resistant starch indicates that dietary interventions may need to be personalised( Reference Walker, Ince and Duncan 57 ).
In another study, a gluten-free diet intervention featuring a reduction in overall polysaccharide intake led to reductions in gut bacterial populations such as Bifidobacterium, Clostridium lituseburense and Faecalibacterium prausnitzii and proportional increases in the abundance of E. coli and total Enterobacteriaceae in healthy volunteers( Reference De Palma, Nadal and Collado 70 , Reference Sanz 72 ).
Based on the available data, differences in the compositions of the gastrointestinal microbiota are demonstrable between groups of people living on different diets. These diet-associated changes in composition can lead to changes in the metabolic activity of the intestinal microbiota, which, in turn, may provoke changes in inflammatory and immune responses. Although attempts to change the composition of the intestinal microbiota by varying the diet have been successful in mice, there is a relative paucity of human dietary intervention studies, and those available are small in sample size and have been conducted over a short period of time. Moreover, mechanisms that link dietary changes to microbial composition alterations remain poorly defined and need to be investigated further. Large, well-controlled trials are also required to determine the impact of altering long-term dietary patterns on the human intestinal microbiota and to elucidate the implications of the key population changes for health and disease.
Antibiotics
Antibiotic treatment has been shown( Reference Claesson, Cusack and O'Sullivan 42 , Reference Woodmansey, McMurdo and Macfarlane 73 – Reference Young and Schmidt 75 ) to dramatically disturb the composition of the faecal microbiota in humans. Palmer et al. ( Reference Palmer, Bik and DiGiulio 76 ) reported changes in the density or composition of the intestinal microbiota in infants following antibiotic treatment. Striking changes have been found in some cases, even to a point where the faecal microbiota was undetectable. As there is considerable inter-individual variability in the composition of the microbiota among humans( Reference Claesson, Cusack and O'Sullivan 42 ), it has been suggested that the impact of antibiotics is best assessed on an individual basis( Reference Jernberg, Löfmark and Edlund 77 ). In general, antibiotic treatment leads to a decrease in the diversity of the microbiota( Reference Jernberg, Löfmark and Edlund 78 ). Nonetheless, the community is quite resilient and can resemble the pre-treatment state in a matter of days or weeks( Reference Claesson, Cusack and O'Sullivan 42 , Reference Dethlefsen, Huse and Sogin 79 , Reference De La Cochetiere, Durand and Lepage 80 ). However, a number of other studies have shown that microbial composition alterations following antibiotic administration can often persist for a long time period following withdrawal of the treatment, with some members of the microbial community failing to return to pre-treatment levels and these may even be lost from the community indefinitely( Reference Jernberg, Löfmark and Edlund 77 , Reference Dethlefsen, Huse and Sogin 79 , Reference Dethlefsen and Relman 81 – Reference Croswell, Amir and Teggatz 83 ). Disruption of the microbiota by antibiotics can also affect the metabolic activity of the bacterial community in the gut. Antibiotic treatment in mice has been shown to drastically alter the intestinal metabolome by affecting host metabolic pathways such as sugar, nucleotide and fatty acid metabolism as well as bile acid, eicosanoid and steroid hormone synthesis coding capacity( Reference Antunes, Han and Ferreira 84 ).
The effect of antibiotics on the intestinal microbiota in infancy is of particular concern. Recent reports have demonstrated that short-term parenteral antibiotic treatment of neonates causes significant alterations in the composition of the gut microbiota including a disturbance of the expected colonisation pattern of bifidobacteria( Reference Fouhy, Guinane and Hussey 85 , 86 ). Colonisation of the intestine early in life has an important role in directing immune system development, and antibiotic use may increase the risk of atopy and allergic asthma by reducing the protective effect of microbial exposure( Reference Russell, Gold and Hartmann 87 , Reference Foliaki, Pearce and Björkstén 88 ). In a large, multi-centre study, Foliaki et al. ( Reference Foliaki, Pearce and Björkstén 88 ) found an association between antibiotic use in the first year of life and symptoms of asthma, rhinoconjunctivitis and eczema in children aged 6 and 7 years.
The impact of antibiotic use on the intestinal bacteria of the elderly is also of interest, since, compared with younger adults, cohorts of elderly populations are typically administered a complex array of medications, including antibiotics. Antibiotic treatment in hospitalised elderly subjects has been shown to increase the intestinal abundance of proteolytic bacteria( Reference Woodmansey, McMurdo and Macfarlane 73 ), to reduce overall bacterial numbers and, in some subjects, to completely eliminate certain bacterial communities( Reference Bartosch, Fite and Macfarlane 46 ). A more recent study has found that antibiotic treatment led to a decrease in the taxonomic richness, diversity and evenness of the intestinal community in elderly subjects, although the magnitude of the changes and the taxa affected were different between subjects. Moreover, the overall community structure was restored within 4 weeks of treatment( Reference Claesson, Cusack and O'Sullivan 42 ).
One of the best-known complications arising following antibiotic therapy is antibiotic-associated diarrhoea (AAD)( Reference Sekirov, Russell and Antunes 89 ). A number of mechanisms underlie the development of AAD. Antibiotic therapy can disturb the natural microbiota in the GIT, which may result in the pathological overgrowth of C. difficile, and it may also disturb the metabolism of carbohydrates, giving rise to maladsorption of osmotically active particles( Reference Young and Schmidt 75 , Reference Vanderhoof, Whitney and Antonson 90 ). Young & Schmidt( Reference Young and Schmidt 75 ) found that in a patient who developed AAD, antibiotic administration was associated with distinct changes in the diversity of the gut microbiota, including a decrease in the prevalence of butyrate-producing bacteria. Following discontinuation of antibiotic treatment, resolution of diarrhoea was accompanied by a reversal of these changes. This provided evidence linking changes in the community structure of the gastrointestinal bacteria with the development of AAD.
The impact of antibiotic use in the short and long terms needs to be investigated further. Longitudinal type studies rather than cross-sectional studies will allow more direct testing of questions regarding the influence of antibiotic use on the development of allergy and gastrointestinal diseases, particularly in early life.
Disease
Inflammatory bowel diseases
Inflammatory bowel diseases (IBD), including Crohn's disease (CD) and ulcerative colitis (UC), are chronic intestinal disorders whose aetiology is unclear. However, an abnormal immune response against luminal antigens, such as dietary factors and/or bacteria, may be involved( Reference Ojetti, Gigante and Ainora 91 , Reference Isaacs and Herfarth 92 ). CD can affect any part of the GIT, although the lower ileum and colon are most commonly involved( Reference Reiff and Kelly 93 ). It is characterised by discontinuous inflammation of the epithelial lining and deep ulcers. UC affects only the colon and rectum and is characterised by continuous mucosal inflammation and superficial ulcers( Reference Gerritsen, Smidt and Rijkers 94 ). The clinical symptoms of IBD include abdominal pain, diarrhoea, rectal bleeding, malaise and weight loss( Reference Reiff and Kelly 93 ).
Numerous studies have compared the compositions of the intestinal microbiota of IBD patients and healthy individuals, and it appears that the dominant microbiota differs between the two groups (reviewed in Ojetti et al. ( Reference Ojetti, Gigante and Ainora 91 ) , Gerritsen et al. ( Reference Gerritsen, Smidt and Rijkers 94 ), Dicksved et al. ( Reference Dicksved, Willing and de Bruijn 95 ) and Shanahan( Reference Shanahan 96 )). Similarly, the dominant microbiota in patients with UC differs from that in patients with CD( Reference Ojetti, Gigante and Ainora 91 , Reference Gerritsen, Smidt and Rijkers 94 ). However, some changes in the composition of the microbiota are similar between the UC and CD patients( Reference Dicksved, Willing and de Bruijn 95 ).
Although the phylum-level changes observed in IBD patients have not been consistent always, in general, an overall decrease in microbial diversity and stability of the intestinal microbiota has been observed in IBD patients( Reference Gerritsen, Smidt and Rijkers 94 , Reference Dicksved, Willing and de Bruijn 95 ). A decrease in the abundance of specific members of the phylum Firmicutes has been reported, which in some cases coincided with an increase in the abundance of those of the phylum Bacteroidetes and that of facultative anaerobes such as Enterobacteriaceae( Reference Gerritsen, Smidt and Rijkers 94 ). Moreover, increased numbers of E. coli, some of which may be pathogenic, have been observed in IBD patients( Reference Dicksved, Willing and de Bruijn 95 ). Increased detection of C. difficile in relapse and remission of both forms of IBD has been observed( Reference Shanahan 96 ). Other reports have described alterations in the abundance of Bacteroidetes spp., proteobacteria, bifidobacteria and lactobacilli, but results have been inconsistent( Reference Dicksved, Willing and de Bruijn 95 ).
With regard to CD, a number of consistent observations have been reported( Reference Shanahan 96 ). These include increased mucosal bacterial counts, increased levels of adherent-invasive E. coli and increased levels of Mycobacterium avium subsp. paratuberculosis. Furthermore, a reduced number of bacteria in the Clostridium leptum group, including F. prausnitzii, have been observed in CD patients( Reference Sokol, Pigneur and Watterlot 97 ). In fact, F. prausnitzii has even been proposed as a potential probiotic for counterbalancing dysbiosis in CD( Reference Sokol, Pigneur and Watterlot 97 ). For UC, a reduced presence of the Clostridium coccoides group has been described, but no specific members of this group have been reported to be associated with the disease yet( Reference Dicksved, Willing and de Bruijn 95 ).
Although marked alterations occur in the composition of the gut microbiota of IBD patients, it is unclear whether these shifts cause the disease or whether they arise due to the changes in the gut environment that result from the disease. Indeed, most of the studies carried out to date have reported associations between the microbiota and IBD only after the IBD phenotype has emerged, which does not allow one to answer the important question of what came first – IBD or a change in the microbiome. More long-term longitudinal studies are needed to examine the progression of diseases and to typify the taxonomic and functional composition changes of the microbiome that lead to or may even define IBD.
Irritable bowel syndrome
Irritable bowel syndrome (IBS) is a common, debilitating gastrointestinal disorder characterised by abdominal pain, bloating and disturbances in bowel function( Reference Malinen, Rinttilä and Kajander 98 – Reference Jeffery, O'Toole and Öhman 100 ). IBS can present as diarrhoea-predominant IBS, constipation-predominant IBS or mixed-bowel-habit IBS( Reference Carroll, Ringel-Kulka and Keku 101 ).
IBS can be difficult to diagnose due to the lack of a biological or pathogenic marker( Reference Malinen, Rinttilä and Kajander 98 , Reference Madden 99 ). Although the pathophysiology of IBS is still not well understood, several factors are thought to play a role. These include malfermentation of food ingredients, altered microbial composition, intestinal motor and sensory dysfunction, immune mechanisms, psychological factors and brain–gut axis dysregulation( Reference Madden 99 , Reference Andresen and Baumgart 102 , Reference Quigley 103 ). Considerable evidence suggests that factors that disturb the gut microbiota, such as gastroenteritis, may contribute to the development of IBS( Reference Thabane, Kottachchi and Marshall 104 ).
Differences in the compositions of the intestinal microbiota between IBS patients and healthy controls (HC) have mostly been studied using faecal material. Mättö et al. ( Reference Mättö, Maunuksela and Kajander 105 ), using culture-based techniques, observed slightly higher numbers of culturable coliforms and an increased aerobe:anaerobe ratio in IBS subjects relative to HC. Moreover, PCR-denaturing gradient gel electrophoresis has revealed more temporal instability in the predominant bacterial population of IBS subjects than in controls, and IBS subjects had more Clostridium spp. and less Eubacterium spp. amplicons. However, the researchers did not control for antibiotic use, which may have contributed to the apparent temporal instability observed( Reference Mättö, Maunuksela and Kajander 105 ). In a subsequent study, which targeted the clostridial groups in IBS, it has been reported that a similar instability existed( Reference Maukonen, Satokari and Mättö 106 ). In addition, a study employing denaturing gradient gel electrophoresis techniques has found that there was significantly more variation in the gut microbiota of healthy volunteers than in that of IBS patients( Reference Codling, O'Mahony and Shanahan 107 ).
Jeffery et al. ( Reference Jeffery, O'Toole and Öhman 100 ) described a detailed analysis of the faecal microbiota in a cohort of well-characterised IBS patients and control subjects and found no uniform change in the composition of the microbiota in IBS patients. However, analysis of the microbial populations revealed distinct clusters, one of which showed normal-like microbial composition compared with HC samples. The other IBS samples were characterised by an increased Firmicutes: Bacteroidetes ratio. In addition, analysis of the IBS microbiota and separate analyses of the two subgroups have shown microbial associations with colonic transit time, satiety, bloating, rectal pain threshold and depression( Reference Jeffery, O'Toole and Öhman 100 ). Significantly, IBS subjects with a microbiota similar to that of the matched HC displayed higher anxiety and depression scores, suggesting a non-intestinal or at least a non-microbiota aetiology for IBS in this subgroup.
A more recent study has shown intestinal dysbiosis in diarrhoea-predominant IBS patients compared with HC. A significant increase in the abundance of unclassified Enterobacteriaceae members and significant reductions in that of the members of the Faecalibacterium genus have been found in IBS patients compared with controls. Furthermore, Enterococcus, Fusobacterium, Pediococcus, unclassified Lactobacillaceae and Veillonella species have been found in IBS patients, but reported to be below detection limits in HC( Reference Carroll, Ringel Kulka and Siddle 108 ).
The studies described above demonstrate that the intestinal microbiota of patients with IBS can differ from that of healthy individuals. Nonetheless, it is not yet possible to be certain (as in IBD discussed above) whether the alterations in the composition of intestinal microflora observed in IBS patients are the cause of IBS or simply a result of the disrupted gut motility or other physiological features of IBS. More studies are needed to clarify whether the microbiota has a causal role in the initiation and/or progression of IBS.
Obesity
Some of the earliest evidence showing the role of the gut microbiota in the regulation of fat storage has been demonstrated in animal models. A pioneering study by Bäckhed et al. ( Reference Bäckhed, Ding and Wang 109 ) has found that GF mice were leaner than their conventional counterparts and colonisation with an intestinal microbiota resulted in a significant increase in body fat content despite lower food consumption in the colonised animals. A subsequent study has found that GF mice were protected against obesity following consumption of a Western-style, high-fat, sugar-rich diet( Reference Bäckhed, Manchester and Semenkovich 110 ). In addition, the colonisation of GF mice with an ‘obese microbiota’ (i.e. from an obese animal) has been reported to lead to greater increases in total body fat compared with GF mice colonised with a ‘lean microbiota’( Reference Turnbaugh, Ley and Mahowald 111 ), indicating that the obese microbiota has an increased capacity to harvest energy from the diet.
It has been suggested that inflammation( Reference Cani, Amar and Iglesias 112 ) and alterations in host gene expression( Reference Bäckhed, Manchester and Semenkovich 110 ) are other mechanisms by which the gut microbiota may influence the host. Obesity and its related metabolic disorder, type 2 diabetes, are generally associated with chronic low-grade inflammation( Reference Wellen and Hotamisligil 113 ). Lipopolysaccharide (LPS), a highly pro-inflammatory component, is a possible initiator of metabolic impairment( Reference Cani, Amar and Iglesias 112 ). Plasma LPS levels increase with higher fat intake in both mice( Reference Cani, Amar and Iglesias 112 ) and humans( Reference Erridge, Attina and Spickett 114 ), and the direct infusion of LPS mimics the physiological effects of a high-fat diet in mice( Reference Cani, Amar and Iglesias 112 ). It has been hypothesised that LPS is taken up with dietary fats in chylomicrons( Reference Ghoshal, Witta and Zhong 115 ) or that LPS reaches the circulation because the gut is more permeable in obese mice due to the disruption of tight junction proteins( Reference Brun, Castagliuolo and Leo 116 , Reference Cani, Possemiers and Van de Wiele 117 ). A review on this topic has been described recently( Reference Sommer and Bäckhed 118 ).
Studies have also linked alterations in the composition of the intestinal microbiota to obesity( Reference Duncan, Lobley and Holtrop 119 – Reference Zhang, DiBaise and Zuccolo 122 ). An increased ratio of Firmicutes:Bacteroidetes has been observed in genetically obese mice (ob/ob)( Reference Ley, Bäckhed and Turnbaugh 120 ) as well as obese humans( Reference Turnbaugh, Hamady and Yatsunenko 23 , Reference Ley, Turnbaugh and Klein 121 ). However, a number of other studies have failed to confirm these findings and have shown variable patterns in phylum-level changes measured in the composition of the microbiota of obese humans( Reference Duncan, Lobley and Holtrop 119 , Reference Schwiertz, Taras and Schafer 123 , Reference Duncan, Belenguer and Holtrop 124 ).
Although it is clear from the studies described above that the gut microbiota is likely to play some role in obesity and metabolic disease, it is difficult to draw definite conclusions on the importance of particular bacterial groups. Further well-designed, large clinical studies are required to identify microbiota-related biomarkers of risk for obesity and metabolic dysregulation.
Manipulation of intestinal microbiota
Probiotics
The term ‘probiotic’, a word derived from Greek and meaning ‘for life’( Reference Quigley 103 ), has been defined as ‘live microorganisms which when administered in adequate amount confer a health benefit on the host’( 125 ). Some probiotic products contain a single strain, while others contain a mixture of several species of bacteria or fungi. The most-studied and commonly used organisms in probiotic preparations are lactobacilli and bifidobacteria.
One of the original concepts associated with probiotics was that their consumption would alter the composition of the intestinal microbiota from a possibly harmful one towards a microbiota that would benefit the host (i.e. replace ‘bad’ bacteria with ‘good’ bacteria)( Reference Metchnikoff 126 ). This was a rather simplistic theory, and it was not based on a full understanding of the complexity of the intestinal microbiota. It has since been suggested( Reference Ouwehand, Salminen and Isolauri 127 ) that too much emphasis should be placed not on the potential change in the composition of the microbiota, but rather on the inherent health benefits conferred by the probiotics themselves. Indeed, for some probiotic effects (e.g. immune modulation), it may not be necessary to achieve a measurable modification of the composition of the intestinal microbiota. In recent times, two main motives have emerged for the use of probiotics. The first is the use of probiotics by healthy subjects to maintain a healthy state and decrease the risk of illness. The second is the use of probiotics as a treatment/therapeutic modality targeted at particular diseases.
There are a variety of proposed health effects (both direct and indirect) of probiotics, which have been reviewed extensively( Reference Parvez, Malik and Ah Kang 128 – Reference Goldin, Kneifel and Salminen 130 ). However, the subtleties of the positive effects of probiotics can only be fully appreciated following a meta-analysis. Some of the most robust clinical data are confined to the preventive and therapeutic effects of probiotic strains on diarrhoeal illness( Reference Videlock and Cremonini 131 – 134 ). A number of beneficial effects of probiotics dealing with intestinal health have been evaluated in Cochrane reviews( 132 , 134 – 136 ). These and other meta-analyses have demonstrated the efficacy of probiotics in the prevention and treatment of AAD( Reference Videlock and Cremonini 131 , Reference Hempel, Newberry and Maher 133 , 136 ), acute infectious diarrhoea( 132 ) and persistent diarrhoea in children( 134 ). Evidence is also accumulating from well-conducted clinical studies on the efficacy of probiotics in the prevention and reduction of the severity of necrotising enterocolitis in premature infants and those with very low birth weight( Reference AlFaleh, Anabrees and Bassler 135 , Reference Barclay, Stenson and Simpson 137 , Reference Wang, Dong and Zhu 138 ). Probiotics have also yielded promising improvements in the prevention and treatment of IBD (UC and CD)( Reference Isaacs and Herfarth 92 ) and IBS( Reference Quigley 103 ). However, it must be noted that these meta-analyses have their own limitations. The clinical and methodological heterogeneity between studies as well as the differences in probiotic type, delivery method (yogurt v. capsule) and dosage makes comparisons difficult. Indeed, no two different probiotics are likely to be functionally the same, and therefore performing meta-analyses based on studies involving different strains, species and even genera is inherently questionable. Different strains may have vastly different effects, and hence no ideal probiotic strain for any of the above-mentioned conditions has been identified, despite continuing advances in this area.
Although there is no single mechanism of action for probiotics, there are a number of common mechanisms by which probiotics might influence the intestinal microbiota (Fig. 2) ( 139 ). However, it is likely that the mechanism of action of probiotics is multifactorial and strain specific( Reference Tuohy, Probert and Smejkal 140 ).
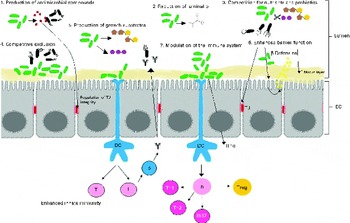
Fig. 2 Schematic diagram illustrating the selected mechanisms by which probiotic bacteria may influence the intestinal microbiota and/or induce beneficial host responses: (1) production of antimicrobial compounds (e.g. bacteriocins); (2) reduction of luminal pH through the production of SCFA; (3) competition with pathogens for nutrients and prebiotics; (4) competitive exclusion of pathogens for adhesion to epithelial cells; (5) production of growth substrates (e.g. vitamins, SCFA and exopolysaccharide); (6) enhanced intestinal barrier function (e.g. increased mucus and β-defensin secretion and/or modulation of cytoskeletal and tight junction protein phosphorylation); (7) modulation of immune response. IEC, intestinal epithelial cells; DC, dendritic cells; TJ, tight junction (modified from O'Toole & Cooney( 139 )). B, B cells; T, T cells; Th, T helper cells; Tn, naive T cells; Treg, regulatory T cells.
While there are extensive scientific and clinical portfolios associated with (specific strains of) probiotics, the European Food Safety Authority is yet to approve health claims for a single probiotic (there have been 120 negative opinions on probiotic claims through February 2011)( Reference Sanders, Heimbach and Pot 141 ). Indeed, regulators are almost applying pharmaceutical standards to the use of probiotics. As has been mentioned already, there are two main uses for probiotics: (1) probiotics as a ‘food-for-health’ product and (2) probiotics as a therapeutic modality for illness. The first example is clearly the one where the food industry is focusing its resources – that is, keeping healthy people healthy. One may ask whether it is appropriate for the regulators to apply pharmaceutical industry standards of proof for probiotics that are largely intended to be given to healthy people? Moreover, unlike pharmaceutical drugs with a single active entity, probiotics encompass hundreds of different strains and hundreds of different surface molecules and possibly metabolites that may be responsible for the ‘probiotic’ effect. Certainly, this is a regulatory challenge that is yet to be resolved. As a scientist interested in exploring the functions of probiotics, one has to be in favour of stringent regulations, and the challenge is now firmly on commercial probiotic purveyors to generate high-quality scientific data that will allow them to make health claims. However, one can also conclude that trying to apply concepts and standards the same as those employed for pharmaceuticals may not be appropriate for probiotics. Scientists, companies and regulators need to address these issues. Otherwise, credibility with the consumer, interest shown by the food industry and ultimately scientific research will be seriously damaged.
Recently, there has been interest in faecal transplantation as an alternative approach for the manipulation of the intestinal microbiota. Indeed, evidence for its use as a treatment for gastrointestinal illness (including pseudomembranous colitis, C. difficile-associated diarrhoea, antibiotic-associated diarrhoea, IBS and IBD) is rapidly accumulating and has been reviewed recently( Reference Anderson, Edney and Whelan 142 – Reference Landy, Al-Hassi and McLaughlin 144 ). Faecal transplantation as a treatment modality remains a controversial issue, and the evidence available for its efficacy is limited. Nonetheless, this therapy holds great promise and further studies are necessary to explore this potential.
Probiotics: dead or alive?
It has been proposed that the minimum therapeutic dose for probiotics is 108–109 viable cells per d( Reference Kailasapathy and Chin 145 ). However, live cells in probiotic products will inevitably lose viability and the actual products will contain varying proportions of populations of viable-to-non-viable/dead cells( Reference Shah, Lankaputhra and Britz 146 ). There may be further losses of viability of the organisms on passage through the relatively hostile environment of the stomach and small intestine( Reference Adams 147 ). Concerns have also been raised that the administration of live micro-organisms may not be appropriate for some population groups (e.g. premature infants and immunocompromised individuals), as they may translocate to the locally draining tissues, thereby causing bacteraemia and sepsis( Reference Kataria, Li and Wynn 148 , Reference Lopez, Li and Kataria 149 ).
Therefore, an area of related ongoing debate is whether or not non-viable forms of beneficial bacterial strains have a role in the conferment of benefits on the host. Indeed, a considerable amount of published scientific evidence indicates that inactivated microbes may positively affect health by influencing the host immune system (reviewed in Adams( Reference Adams 147 ), Kataria et al. ( Reference Kataria, Li and Wynn 148 ) and Taverniti & Guglielmetti( Reference Taverniti and Guglielmetti 150 )). The ability of bacterial cells to potentially interact with the host, independent of viability, is based on the capacity of human cells to recognise specific bacterial components or products, leading to responses that commonly involve the mucosa-associated lymphoid tissue and, therefore, the immune system( Reference Adams 147 ). Some studies have proposed that the immunomodulatory effects exerted by non-viable probiotics may be due to their immunostimulatory DNA, cell wall components, peptidoglycan, intra- and extracellular polysaccharide products and cell-free extracts( Reference Lammers, Brigidi and Vitali 151 – Reference Dalpke, Frey and Morath 154 ).
A number of studies have evaluated the immunomodulatory effect of the probiotic Lactobacillus rhamnosus GG, in both live and dead (inactivated) forms. Heat-killed or UV-inactivated L. rhamnosus GG may reduce inflammation by decreasing experimentally induced IL8 production by epithelial Caco-2 cells( Reference Lopez, Li and Kataria 149 , Reference Zhang, Li and Caicedo 155 ). In the absence of induction, high doses of live L. rhamnosus GG actually induce IL8 production, while the heat-killed agent causes only a slight increase in IL8 production, meaning that it has a lower potential to cause inflammation itself( Reference Zhang, Li and Caicedo 155 ), thus indicating that the heat-killed agent may be a safer alternative. A similar response has been demonstrated in an animal model, in which both live and heat-killed L. rhamnosus GG reduced the levels of LPS-induced pro-inflammatory mediators and up-regulated the levels of anti-inflammatory mediators in intestinal tissue in rats( Reference Li, Russell and Douglas-Escobar 156 ). Similarly, heat-killed Lactobacillus strains have been found to induce TNFα secretion in mouse splenic mononuclear cells to various degrees( Reference Matsuguchi, Takagi and Matsuzaki 157 ). Furthermore, the purified surface glycolipid lipoteichoic acid, which is a major component of the cell wall of lactobacilli, activated macrophages through toll-like receptor 2 (TLR2) in a strain-specific manner. It has even been suggested that the immense structural diversity in lipoteichoic acid derived from different bacteria may induce a variety of immunoregulatory effects( Reference Ciszek-Lenda, Nowak and Śróttek 158 ).
The immunoregulatory potential of exopolysaccharide (EPS) has also been investigated( Reference Ciszek-Lenda, Nowak and Śróttek 158 – Reference López, Monteserín and Gueimonde 162 ). It has been shown that in vitro levels of pro-inflammatory cytokines are highly elevated upon exposure of mouse splenocytes to cells of EPS-deficient Bifidobacterium breve co-cultures, whereas exposure to an EPS-producing strain has been shown to markedly reduce the levels of these cytokines. Moreover, treatment of mice with EPS+ B. breve has been shown to elicit reduced levels of pro-inflammatory immune cells compared with EPS− strains( Reference Fanning, Hall and Cronin 163 ). A recent study has described the stimulatory effects of a Lactobacillus-derived EPS on the release of inflammatory mediators by mouse peritoneal macrophages in vitro ( Reference Ciszek-Lenda, Nowak and Śróttek 158 ). EPS effectively induced the production of mediators and cytokines by macrophages, especially TNFα, IL6 and IL12. Interestingly, EPS induced higher levels of TNFα and IL6 than of IL10, suggesting a net pro-inflammatory potential. However, its stimulatory effect was significantly lower than that of LPS or whole, killed bacterial cells. Moreover, whole cells were stronger inducers of anti-inflammatory IL10 than EPS alone, suggesting that intact bacteria and EPS may have an opposing effect on macrophage polarisation( Reference Ciszek-Lenda, Nowak and Śróttek 158 ). Similarly, Wu et al. ( Reference Wu, Pan and Wu 159 ) investigated the effect of heat-killed Bifidobacterium longum and its isolated EPS fraction on the activities of a murine macrophage cell line, including induction of IL10 and TNFα production. EPS exposure stimulated growth and induced IL10 secretion in macrophages as well as induced lower levels of TNFα secretion. LPS, on the other hand, induced high levels of TNFα secretion and EPS pre-treatment prevented LPS-induced release of TNFα. As both EPS and LPS are surface macromolecules with oligosaccharide moieties, the authors concluded that EPS may act as a LPS blocker. Although EPS may play a role in immune regulation, information about the molecular mechanisms by which EPS interacts with the immune system is scarce( 161 ). Defining the common biological properties of EPS has proved to be difficult because of its enormous structural diversity( Reference Ciszek-Lenda, Nowak and Śróttek 158 ).
Some studies have found that bacterial DNA may be partly responsible for the immunomodulatory effect of probiotics. The administration of non-viable, irradiated probiotics (VSL#3) but not heat-killed probiotics has been shown to effectively ameliorate experimental colitis in mice mediated by a TLR9–probiotic DNA interaction( Reference Rachmilewitz, Katakura and Karmeli 152 ). Similarly, Bifidobacterium genomic DNA has been shown to induce the secretion of the anti-inflammatory IL10 by human peripheral blood mononuclear cells( Reference Lammers, Brigidi and Vitali 151 ).
Immunoactive components of probiotic bacteria may not be limited to cell wall structures and DNA. There have been reports concerning a soluble immunomodulator in bifidobacteria. The immunomodulating activity of Bifidobacterium adolescentis increases after disruption of the cells by sonication and the immunopotentiating activity appears in the soluble fraction following centrifugation, indicating the existence of an intracellular soluble immunomodulator( Reference Hosono, Lee and Ametani 153 ). The components of Bifidobacterium pseudocatenulatum also have immunomodulatory effects, which appear to be dependent on the method of preparation( Reference Hiramatsu, Hosono and Takahashi 164 ). Compared with heat-treated and untreated cells, sonicated Bifidobacterium has been shown to be the most potent inducer of innate immune responses in Peyer's patch cells in vitro and in vivo (following a single-shot oral administration to mice). However, heat-treated Bifidobacterium has been shown to exhibit the greatest immunomodulatory activity following repeated oral administration (for seven consecutive days) in mice. The researchers concluded that the immunomodulatory effect of Bifidobacterium is dependent upon the bacterial conformation and condition( Reference Hiramatsu, Hosono and Takahashi 164 ).
Although there is substantial evidence from in vitro and animal studies that inactivated probiotics can act as biological response modifiers, there is a relative paucity of information on the effect of dead probiotics in vivo in human clinical trials. However, a number of basic human consumption studies have been carried out with Lacteol Fort (Lactobacillus acidophilus LB that is heat-killed and freeze-dried in the presence of its fermented culture medium)( Reference Xiao, Zhang and Lu 165 , Reference Halpern, Prindiville and Blankenburg 166 ). Lacteol Fort has been shown to improve the clinical symptoms (by decreasing bowel movement, abdominal pain and distension and by improving stool consistency and the feeling of incomplete evacuation) of chronic diarrhoea( Reference Xiao, Zhang and Lu 165 ), possibly through a mechanism involving competitive exclusion( Reference Chauvière, Coconnier and Kerneis 167 ).
In conclusion, while a number of studies have proposed that the viability of probiotics is not essential to exert an immunomodulatory effect, this is not a uniform feature of all probiotics tested to date, as different in vitro studies have also reported that a viable probiotic is essential to exert an immunomodulatory effect( Reference Arribas, Garrido-Mesa and Perán 168 , Reference Ma, Forsythe and Bienenstock 169 ). In addition, the method of preparation may play a significant role as the immunomodulatory effect may be dependent on the bacterial conformation and condition, as has been described above( Reference Rachmilewitz, Katakura and Karmeli 152 , Reference Hiramatsu, Hosono and Takahashi 164 ). Further study is essential to elucidate whether inactivated probiotic bacteria or their products are able to exert beneficial effects similar to those exerted by live bacteria in vivo. Work on the specific mechanisms is also required to explain what is actually being triggered by the dead agents and whether this is similar to the mechanisms of live agents( Reference Li, Russell and Douglas-Escobar 156 ). Biological products based on dead cells might be relatively easy to produce, commercialise and standardise and would have the added advantage of having a much longer shelf life. In addition, use of dead probiotics may be, in some circumstances, safer than using live probiotics. Resolution of this debate might also require participation of relevant stakeholders in re-examining the definition of a probiotic, which is currently restricted to live cells( 125 ). It must be noted that while the viability of probiotics may not be necessary to exert an immunomodulatory effect, a number of mechanisms mediating the health benefits of probiotics do require viability, such as the metabolism of non-digestible polysaccharides and production of metabolites (e.g. SCFA). Hence, one should be cautious while applying the term ‘probiotic’ to these dead cell preparations.
Conclusion
The intestinal microbiota is undoubtedly an important factor in determining the health status of the host and has been implicated in both gastrointestinal and extra-intestinal disorders. The studies described in the present review raise the question of whether it is now possible to deduce the composition of a ‘healthy’ normal microbiota. Indeed, limited accessibility of the different parts of the GIT (including the colonic mucosa) as well as the individual-specific, complex composition of the GIT microbiota makes our understanding of this community somewhat incomplete( Reference Zoetendal, Rajilic-Stojanovic and de Vos 170 ). It is also essential to bear in mind the influence of different dietary patterns on the activity and composition of the microbiota and the potential implications for host health. Although several large international studies are striving to improve our knowledge of the composition of the gut biota, an overwhelming majority of microbes that compose these microbial communities are not yet characterised in detail to any great level( Reference Dusko Ehrlich 5 ). Comparison of data from different studies is difficult as sample processing and analysis methods vary between research groups. Moreover, our apparent inability to culture all members of the microbiota makes it currently impossible to create hypotheses regarding the role of these uncultured microbes in health and disease( Reference Zoetendal, Rajilic-Stojanovic and de Vos 170 ). As we are still unable to completely define the microbiota of a healthy intestinal tract, it is similarly difficult to define the microbiota associated with an intestinal disorder. However, major advances in human metagenomics have now provided a catalogue of 3·3 million non-redundant genes( Reference Dusko Ehrlich 5 , Reference Qin, Li and Raes 8 ). This catalogue will enable the development of gene profiling approaches that aim to detect associations of bacterial genes and phenotypes. These developments should lead to rapid advances in diagnostic and prognostic tools as well as pave the way to rational approaches to the manipulation of an individual's intestinal microbiota to promote health.
The improvement or maintenance of health through the use of probiotics has been the focus of extensive research. Indeed, the probiotic market has expanded rapidly in recent years and a large variety of probiotic products are available. However, the efficacy of probiotics is strain and dose dependent, and the clinical and methodological differences (strain, dose and formulation) between studies make comparisons difficult. Hence, there is presently strong evidence to support their use only under a few conditions. It is also acknowledged that in most cases, the exact mechanisms of the beneficial effects are not fully understood. Although many studies have provided information on several possible modes of action, it has not been possible to identify definite cause–effect relationships. Stringent requirements imposed by regulatory authorities such as the European Food Safety Authority require more solid scientific evidence to support any health claims associated with probiotic products. Future research requires well-designed, large, randomised, double-blind placebo-controlled clinical trials along with more mechanistic studies on cell and animal models in order to strengthen our evidence base. Indeed, the development of novel in vitro models of the human intestinal epithelium (e.g. bioreactors and organoids) will increase our understanding of the molecular mechanisms of host–microbe interactions and pave the way for future ventures aimed at bioengineering human intestine( Reference Bermudex-Brito, Plaza-Díaz and Fontana 171 ). Further investigation into the health benefits of ingesting dead organisms in vivo is also required. Indeed, the potential health effect of these non-viable bacteria depends on whether the mechanism of the health effect of probiotics is dependent on viability, and hence each probiotic strain should be assessed on a case-by-case basis. Furthermore, as the term ‘probiotic’ fails to account for the use of dead organisms, it has been suggested that the term ‘pharmabiotic’ would be more inclusive( Reference Shanahan 172 ).
Acknowledgements
The Alimentary Pharmabiotic Centre is a research centre funded by the Science Foundation Ireland (SFI), through the Irish Government's National Development Plan. S. E. P. is supported by the Irish Research Council postgraduate scholarship Enterprise Partnership Scheme (in collaboration with Alimentary Health Limited) and by the Alimentary Pharmabiotic Centre (SFI grant no. 07/CE/B1368). The present review was also supported by the Irish Government's National Development Plan by way of a Department of Agriculture Food and Marine and Health Research Board FHRI award to the ELDERMET project. The above-mentioned funding agencies had no role in the design, analysis or writing of this article.
S. E. P. wrote the manuscript. G. F. F., P. W. O. T., R. P. R. and C. S. critically reviewed the manuscript and contributed to its revision. All authors read, reviewed and approved the final version of the manuscript.
The authors do not have any conflicts of interest.



