There is considerable evidence suggesting that in human subjects, elevated intake of sugar-sweetened beverages or added sugars in the form of sucrose and/or fructose is associated with increased body weight, the presence of unfavourable lipid levels, insulin resistance, fatty liver, type 2 diabetes, CVD and the metabolic syndrome(Reference Stanhope1). Concerning the specific effects of the monosaccharide fructose on components of the metabolic syndrome, authors of recent reviews have concluded that high fructose intakes have substantial, even profound, deleterious metabolic consequences that possibly predispose individuals to chronic conditions such as type 2 diabetes and CVD(Reference Tappy and Le2, Reference Dekker, Su and Baker3).
It has been previously shown that adult sedentary rats fed a high-fructose diet for 8 weeks exhibit hepatic insulin resistance, increased lipogenesis and lipid deposition(Reference Crescenzo, Bianco and Falcone4), in agreement with previous findings(Reference Mayes5–Reference Park, Lemieux and Lewis8) and with the fact that about 90 % of fructose coming from the diet is metabolised in the liver(Reference Tappy and Le2). Increased hepatic lipid synthesis implies higher lipid circulation that could influence other tissues, such as skeletal muscle, and have a deep impact on whole-body metabolic homeostasis. Several data in the literature have underlined a potential link between insulin resistance and mitochondrial derangement in the skeletal muscle of human and animal models(Reference Chaveza and Summers9–Reference Johannsen and Ravussin11), although this notion is challenged by several findings showing no mitochondrial defect with insulin resistance(Reference Boushel, Gnaiger and Schjerling12–Reference Nair, Bigelow and Asmann17). On the basis of these considerations, the effect of long-term fructose feeding on skeletal muscle mitochondrial energetics was investigated. To this end, mitochondrial mass, oxidative capacity and efficiency in skeletal muscle were assessed. Measurements in isolated tissue were coupled with the determination of whole-body RMR and glucose tolerance.
Research methods and procedures
Male Sprague–Dawley rats (Charles River) of 90 d of age were caged singly in a temperature-controlled room (23 ± 1°C) with a 12 h light–12 h dark cycle (06.30–18.30 hours) and divided into two groups with the same initial body weight that were fed a high-fructose or control diet (Mucedola 4RF21; Settimo Milanese) for 8 weeks. The composition of the two diets is given in Tables 1 and 2. Treatment, housing and killing of animals met the guidelines set by the Italian Health Ministry. All experimental procedures involving animals were approved by ‘Comitato etico-scientifico per la sperimentazione animale’ of the University ‘Federico II’ of Naples.
Table 1 Composition of the experimental diets

AIN, American Institute of Nutrition.
* AIN, 1977.
† AIN, 1980.
‡ Estimated by computation using values (kJ/g) for energy content as follows: protein 16·736, lipid 37·656 and carbohydrate 16·736.
Table 2 Micronutrient composition of the experimental diets

At the end of the experimental period, the glucose tolerance test was carried out. Then, the animals were killed by decapitation, the skeletal muscle was harvested and the carcasses were used for body composition determination.
RMR
At the beginning and after 2, 4 6 and 8 weeks of the dietary treatment, VO2 of the rats was recorded with a monitoring system (Panlab s.r.l.) composed of a four-chamber indirect open-circuit calorimeter, designed for the continuous monitoring of up to four rats simultaneously. Measurements were performed every 15 min for 3 min in each cage. Food was withdrawn at 07.00 hours and mean values of a 2 h period (13.00–15.00 hours) were taken as an indicative of post-absorptive RMR.
Glucose tolerance test and plasma NEFA
Rats were fasted for 6 h from 08.00 hours. A point 0 sample was obtained from venous blood from a small tail clip and then glucose (2 g/kg body weight) was injected intraperitoneally. Blood samples were collected after 20, 40, 60, 90, 120 and 150 min. The blood samples were centrifuged at 1400 g av for 8 min at 4°C. Plasma was removed and stored at − 20°C until used for determination of substrates and hormones. Plasma glucose concentration was measured by the colorimetric enzymatic method (Pokler Italia). Plasma insulin concentration was measured using an ELISA kit (Mercodia AB) in a single assay to remove inter-assay variations.
Plasma NEFA concentrations were measured by the colorimetric enzymatic method using a commercial kit (Randox Laboratories Limited).
Body and skeletal muscle composition
Guts were cleaned of undigested food and the carcasses were then autoclaved. After dilution (1:2 in distilled water) and subsequent homogenisation of the carcasses with a Polytron homogeniser (Kinematica), the resulting homogenates were frozen at − 20°C until the day of measurements. Duplicate samples of the homogenised carcasses were analysed for energy content by bomb calorimetry. To take into account the energy content of the harvested skeletal muscle, tissue samples were dried, the energy content was then measured with the bomb calorimeter and added to the body energy content. Total body and skeletal muscle lipid contents were measured by the Folch extraction method(Reference Folch, Lees and Sloane Stanley18). Total body protein content was determined using a formula relating total energy value of the carcass, energy derived from fat and energy derived from protein(Reference Dulloo and Girardier19); the energy values for body fat and protein were taken as 39·2 and 23·5 kJ/g, respectively(Reference Armsby20). Skeletal muscle TAG and cholesterol were measured by the colorimetric enzymatic method using commercial kits (SGM Italia), phospholipids were obtained by subtracting TAG and cholesterol contents from the total lipid content and glycogen was assessed by the direct enzymatic procedure(Reference Roehrig and Allred21). Skeletal muscle ceramide content was evaluated by ELISA(Reference Guilbault, De Sanctis and Wojewodka22) using ninety-six-well Polysorp plates (Nunc). In brief, skeletal muscle lipids (70 μl in methanol) were adsorbed to the well bottoms overnight at 4°C. The plates were blocked with 10 mm-PBS, 140 mm-NaCl and 0·1 % Tween (pH 7·4) supplemented with 1 % bovine serum albumin (BSA) for 1 h at 37°C. The plates were then washed three times with 10 mm-PBS, 140 mm-NaCl and 0·05 % Tween (pH 7·4) (Tween–PBS), and incubated with a monoclonal anti-ceramide antibody (2 μg/ml) for 1 h at 37°C. After three washings in Tween–PBS, peroxidase-conjugated goat anti-mouse IgM (1:5000 dilution) was incubated with the plates for 1 h at 37°C. After four washings in Tween–PBS, the wells were incubated with 100 μl of a colour development solution (20 mg o-phenylenediamine dihydrochloride in 50 ml of 70 mm-Na2HPO4, 30 mm-citric acid, pH 5, supplemented with 120 μl of 3 % H2O2). After 15 min at 37°C, the reaction was stopped by the addition of 50 μl of 2·5 m-H2SO4 and absorbance was measured at 492 nm. All tests were done in triplicate. Immunoreactivity was normalised to initial tissue weight. Negative control reactions included the omission of the primary antibody.
Metabolisable energy intake was determined as detailed previously(Reference Lionetti, Mollica and Crescenzo23), by subtracting the energy measured in the faeces and urine from the gross energy intake, determined from daily food consumption and gross energy density of the diet.
Preparation of skeletal muscle isolated mitochondria and measurements of mitochondrial oxidative capacities, degree of coupling and uncoupling effect of fatty acids
Hind leg muscles were rapidly removed and used for the preparation of isolated mitochondria. Hind leg muscles were freed of excess connective tissue, finely minced, washed in a medium containing 100 mm-KCl, 50 mm-Tris, pH 7·5, 5 mm-MgCl2, 1 mm-EDTA, 5 mm-ethylene glycol tetraacetic acid and 0·1 % (w/v) fatty acid-free BSA, and treated with protease nagarse (1mg/g) for 4 min. Tissue fragments were then homogenised with the above medium (1:8, w/v) at 500 rpm (4 strokes/min). Aliquots of the homogenate were withdrawn for measurements of state 3 mitochondrial respiration, while the rest was centrifuged at 3000 g av for 10 min, the resulting supernatant was rapidly discarded and the pellet was resuspended and centrifuged at 500 g av for 10 min. The supernatant was then centrifuged at 3000 g av for 10 min, the pellet was washed once and resuspended in a suspension medium (250 mm-sucrose, 50 mm-Tris, pH 7·5 and 0·1 % fatty acid-free BSA).
Oxygen consumption rate was measured polarographically with a Clark-type electrode (Yellow Springs Instruments) in a 3 ml glass cell at 30°C. Skeletal muscle homogenates or isolated mitochondria were incubated in a medium containing 30 mm-KCl, 6 mm-MgCl2, 75 mm-sucrose, 1 mm-EDTA, 20 mm-KH2PO4, pH 7·0, and 0·1 % (w/v) fatty acid-free BSA, pH 7·0. The substrates used were 10 mm-succinate+3·8 μm-rotenone, 10 mm-glutamate+2·5 mm-malate, 40 μm-palmitoyl-coenzyme A+2 mm-carnitine+2·5 mm-malate, 40 μm-palmitoyl-carnitine+2·5 mm-malate or 10 mm-pyruvate+2·5 mm-malate. State 3 oxygen consumption was measured in the presence of 0·3 mm-ADP.
The degree of coupling (q) was determined in skeletal muscle mitochondria by applying equation 11 by Cairns et al. (Reference Cairns, Walther and Harken24):
 $$\begin{eqnarray} q = \sqrt {1 - ( J _{o})_{sh}/( J _{o})_{unc}}, \end{eqnarray}$$
$$\begin{eqnarray} q = \sqrt {1 - ( J _{o})_{sh}/( J _{o})_{unc}}, \end{eqnarray}$$where (J o)sh represents the oxygen consumption rate in the presence of oligomycin that inhibits ATP synthase and (J o)unc is the uncoupled rate of oxygen consumption induced by trifluorocarbonylcyanide phenylhydrazone (FCCP), which dissipates the transmitochondrial proton gradient. (J o)sh and (J o)unc were measured as above using succinate 10 mm+rotenone 3·75 μm in the presence of oligomycin (2 μg/ml) or FCCP (1 μm), respectively, both in the absence and presence of palmitate at a concentration of 45 μm.
The uncoupling effect of the fatty acid palmitate was assessed by measuring the mitochondrial membrane potential before and after the addition of increasing concentrations of palmitate. Mitochondrial membrane potential recordings were performed with safranin O using a JASCO dual-wavelength spectrophotometer (511–533 nm). Measurements were made at 30°C in a medium containing 30 mm-LiCl, 6 mm-MgCl2, 75 mm-sucrose, 1 mm-EDTA, 20 mm-Tris-PO4, pH 7·0, and 0·1 % (w/v) fatty acid-free BSA, pH 7·0, in the presence of succinate (10 mm), rotenone (3·75 μm), oligomycin (2 μg/ml) and safranin O (83·3 nmol/mg), both in the absence and after the addition of 15, 30 and 45 μm-palmitate. Absorbance readings were transformed into mV membrane potential using the Nernst equation:
 $$\begin{eqnarray} \Delta \Psi = 61\hairsp mV\cdot log\,([K^{ + }]_{in}/[K^{ + }]_{out}). \end{eqnarray}$$
$$\begin{eqnarray} \Delta \Psi = 61\hairsp mV\cdot log\,([K^{ + }]_{in}/[K^{ + }]_{out}). \end{eqnarray}$$Calibration curves made for each preparation were obtained from traces in which the extramitochondrial K+ level ([K+]out) was altered in the 0·1–20 mm range. The change in absorbance caused by the addition of 3 μm-valinomycin was plotted against [K+]out. Then, [K+]in was estimated by extrapolation of the line to the zero uptake point.
Western blot quantification of skeletal muscle Akt, phosphorylated Akt and mitochondrial cytochrome c
Western blot analyses were carried out as described previously(Reference Crescenzo, Bianco and Falcone25). Briefly, skeletal muscle samples were denatured in a buffer (60·0 mm-Tris, pH 6·8, 10 % sucrose, 2 % SDS and 4 % β-mercaptoethanol) and loaded onto a 12 % SDS–polyacrylamide gel. After the run in electrode buffer (50 mm-Tris, pH 8·3, 384 mm-glycine and 0·1 % SDS), the gels were transferred onto polyvinylidene difluoride membranes (Immobilon-P; Millipore) at 0·8 mA/cm2 for 90 min. The membranes were pre-blocked in blocking buffer (PBS, 5 % milk powder and 0·5 % Tween 20) for 1 h, and then incubated overnight at 4°C with a polyclonal antibody for Akt and phosphorylated (p)-Akt (diluted 1:1000 in blocking buffer; Cell Signaling) or with a mouse monoclonal antibody for cytochrome c (diluted 1:100 in blocking buffer; Biomol International). The membranes were washed three times for 12 min in PBS/0·5 % Tween-20 and three times for 12 min in PBS, and then incubated for 1 h at room temperature with an anti-mouse, alkaline phosphatase-conjugated secondary antibody (Promega). The membranes were washed as described above, rinsed in distilled water and incubated at room temperature with a chemiluminescent substrate (CDP-Star; Sigma-Aldrich). Data detection was carried out by exposing autoradiography films (Kodak; Eastman Kodak Company) to the membranes. Quantification of signals was carried out using Un-Scan-It gel software (Silk Scientific).
Mitochondrial lipid peroxidation and superoxide dismutase specific activity
Lipid peroxidation was determined according to Fernandes et al. (Reference Fernandes, Custodio and Santos26), by measuring thiobarbituric acid-reactive substances. Aliquots of mitochondrial suspensions were added to 0·5 ml of ice-cold 40 % TCA. Then, 2 ml of 0·67 % of aqueous thiobarbituric acid containing 0·01 % of 2,6-di-tert-butyl-p-cresol were added. The mixtures were heated at 90°C for 15 min, then cooled in ice for 10 min and centrifuged at 850 g for 10 min. The supernatant fractions were collected and lipid peroxidation was estimated spectrophotometrically at 530 nm. The amount of thiobarbituric acid-reactive substances formed was calculated using a molar extinction coefficient of 1·56 × 105 per m/cm and expressed as nmol thiobarbituric acid-reactive substances/mg protein. Superoxide dismutase (SOD) specific activity was measured according to Flohè & Otting(Reference Flohè and Otting27). Determinations were carried out spectrophotometrically (550 nm) at 25°C in a medium containing 0·1 mm-EDTA, 2 mm-KCN, 50 mm-KH2PO4, pH 7·8, 20 mm-cytochrome c, 0·1 mm-xanthine and 0·01 units of xanthine oxidase, by monitoring the decrease in the reduction rate of cytochrome c by superoxide radicals, generated by the xanthine–xanthine oxidase system. One unit of SOD activity is defined as the concentration of the enzyme that inhibits cytochrome c reduction by 50 % in the presence of xanthine+xanthine oxidase(Reference Flohè and Otting27).
Chemicals
All chemicals utilised were of analytical grade and were purchased from Sigma.
Statistical analysis
Data are provided as means with their standard errors. Statistical analyses were performed using a two-tailed, unpaired, Student's t test or repeated-measures two-way ANOVA for main effects and interactions followed by Bonferroni's post-test. All analyses were performed using GraphPad Prism 4 (GraphPad Software, Inc.).
Results
Body composition measurements carried out at the end of 8 weeks of the dietary treatment show that body energy was significantly higher in fructose-fed rats than in control rats (Table 3), due to an increase in energy from lipids but not in energy from proteins. Cumulative metabolisable energy intake over the 8 weeks of the dietary treatment was similar between the two groups (control rats 19 600 (sem 944) kJ; fructose-fed rats 19 400 (sem 453) kJ), thus indicating an increased metabolic efficiency in fructose-fed rats. As for the skeletal muscle composition, a significant increase in total lipids, TAG and ceramide was found in fructose-fed rats compared with the controls, while no variation was found in cholesterol, phospholipids and glycogen. Rats fed a fructose-rich diet exhibited a significant increase in plasma NEFA (Table 3).
Table 3 Energetic and metabolic characterisations in rats fed a high-fructose or control diet for 8 weeks (Mean values with their standard errors, n 6 rats per group)

AU, absorbance units.
* Mean values were significantly different compared with the controls (P< 0·05; two-tailed, unpaired Student's t test).
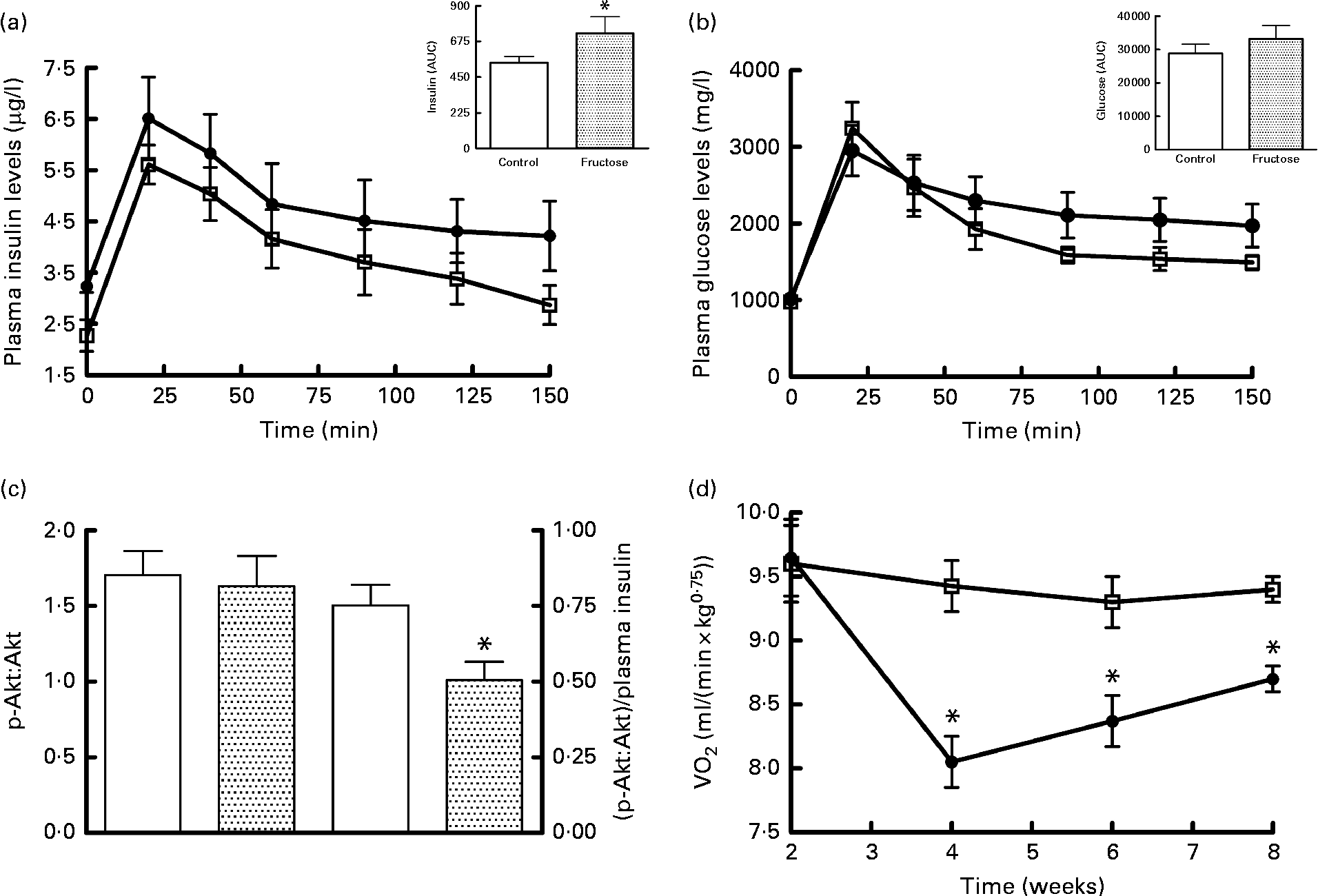
The results of the glucose tolerance test carried out in fructose-fed and control rats at the end of the diet treatment are presented in Fig. 1. The insulin response was significantly higher in fructose-fed rats compared with the controls (Fig. 1(a)), while no significant variation in plasma glucose levels was found, although the values for fructose-fed rats tended to be higher than those for control rats (Fig. 1(b)). Western blot analysis of the p-Akt:Akt ratio in skeletal muscle from fructose-fed and control rats showed no significant variation due to the dietary treatment (Fig. 1(c)). When p-Akt levels were normalised to insulin plasma levels, significantly lower values were found in fructose-fed rats compared with the controls (Fig. 1(c)). Finally, significantly lower RMR values were found in fructose-fed rats compared with the controls, starting from week 4 of the dietary treatment (Fig. 1(d)).

Fig. 1 Plasma (a) insulin and (b) glucose levels after a glucose load (![]() , control;
, control;![]() , fructose), (c) Western blot quantification of phosphorylated (p)-Akt:Akt levels and p-Akt:plasma insulin levels in skeletal muscle and (d) time course of RMR in fructose-fed (
, fructose), (c) Western blot quantification of phosphorylated (p)-Akt:Akt levels and p-Akt:plasma insulin levels in skeletal muscle and (d) time course of RMR in fructose-fed (![]() ) or control (□) rats. Values are means (n 6 rats per group), with their standard errors represented by vertical bars. AUC was calculated using the trapezoid method. * Mean values were significantly different compared with the controls (P< 0·05; two-tailed, unpaired Student's t test for insulin AUC and p-Akt, repeated-measures two-way ANOVA for main effects and interactions followed by Bonferroni's post-test for RMR).
) or control (□) rats. Values are means (n 6 rats per group), with their standard errors represented by vertical bars. AUC was calculated using the trapezoid method. * Mean values were significantly different compared with the controls (P< 0·05; two-tailed, unpaired Student's t test for insulin AUC and p-Akt, repeated-measures two-way ANOVA for main effects and interactions followed by Bonferroni's post-test for RMR).
Mitochondrial oxidative capacities were assessed in isolated mitochondria from skeletal muscle using NAD, FAD and lipid substrates (Table 4) and the results obtained showed no significant variation in fructose-fed rats compared with the controls. Mitochondrial protein mass was assessed by measuring homogenate cytochrome c content. The results reported in Table 4 showed that the cytochrome c:actin ratio was significantly higher in fructose-fed rats compared with the controls. As a consequence, state 3 mitochondrial respiratory rates assessed in whole-tissue homogenates were found to be significantly higher in fructose-fed rats compared with the controls (Table 4).
Table 4 Mitochondrial oxidative capacities, mass and oxidative status in skeletal muscle from rats fed a high-fructose or control diet for 8 weeks (Mean values with their standard errors, n 6 rats per group)

* Mean values were significantly different compared with the controls (P< 0·05; two-tailed, unpaired Student's t test).
Lipid peroxidation and SOD specific activity were measured and taken as an index of cellular oxidative damage and antioxidant defences, respectively (Table 4). A significant increase in lipid peroxidation and a significant decrease in SOD activity were found in fructose-fed rats compared with the controls.
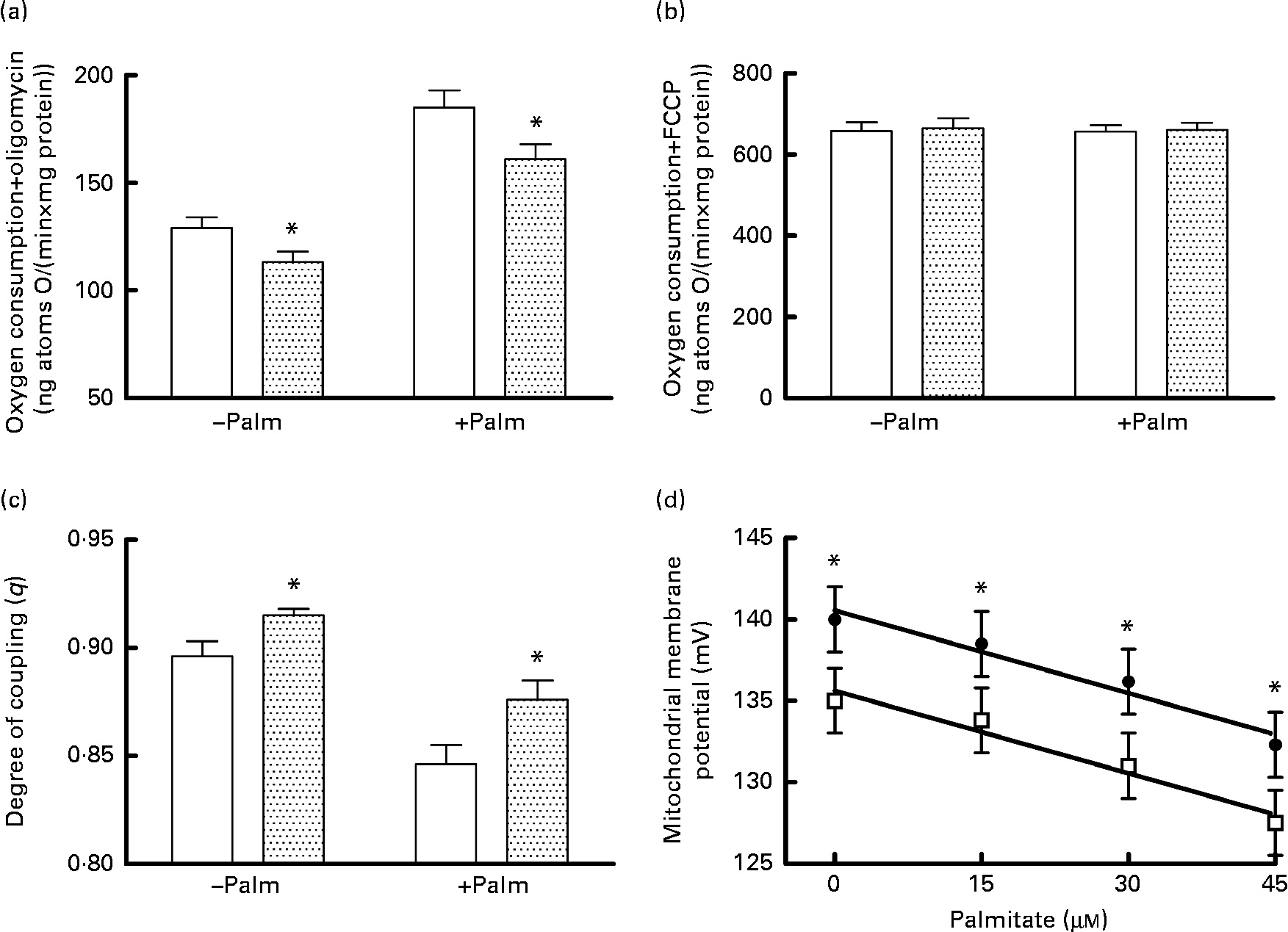
Mitochondrial energetic efficiency was assessed through the measurement of state 4 respiration in the presence of oligomycin and uncoupled respiration in the presence of FCCP, as well as through the evaluation of mitochondrial membrane potential in state 4 conditions, both in the absence and presence of physiological concentrations of palmitate. Oligomycin-induced state 4 respiration was significantly lower, both in the absence and presence of palmitate (Fig. 2(a)), while maximal FCCP-stimulated respiration was not affected by high fructose feeding, both in the absence and presence of palmitate (Fig. 2(b)), so that mitochondrial efficiency, assessed as the degree of coupling (q), was significantly higher in fructose-fed rats compared with the controls, both in the absence and presence of palmitate (Fig. 2(c)). Mitochondria from fructose-fed rats were less responsive to the uncoupling effect of fatty acids, since the mitochondrial membrane potential in state 4 conditions was found to be significantly higher in fructose-fed rats compared with the controls, both in the absence and presence of increasing concentrations of the fatty acid palmitate (Fig. 2(d)).
Fig. 2 Oxygen consumption in the (a) presence of oligomycin (□, control;![]() , fructose) or (b) uncoupled by trifluorocarbonylcyanide phenylhydrazone (□, control;
, fructose) or (b) uncoupled by trifluorocarbonylcyanide phenylhydrazone (□, control; ![]() , fructose), (c) degree of coupling values calculated from oxygen consumption in the presence of oligomycin and uncoupled by FCCP (□, control;
, fructose), (c) degree of coupling values calculated from oxygen consumption in the presence of oligomycin and uncoupled by FCCP (□, control;![]() , fructose) and (d) membrane potential in state 4 conditions in the absence and presence of palmitate in hepatic mitochondria from fructose-fed (●) and control (□) rats. Values are means (n 6 per group), with their standard errors represented by vertical bars. * Mean values were significantly different compared with the controls (P< 0·05; two-way ANOVA for main effects and interactions followed by Bonferroni's post-test).
, fructose) and (d) membrane potential in state 4 conditions in the absence and presence of palmitate in hepatic mitochondria from fructose-fed (●) and control (□) rats. Values are means (n 6 per group), with their standard errors represented by vertical bars. * Mean values were significantly different compared with the controls (P< 0·05; two-way ANOVA for main effects and interactions followed by Bonferroni's post-test).
Discussion
The main result of the present study is that long-term high fructose intake in adult rats induces an increase in skeletal muscle mitochondrial efficiency that could contribute to the condition of energy sparing (reduced RMR) found here.
First, the present results underline an important role for diet composition in the induction of obesity and indicate that not only a high lipid content but also the presence of simple sugars can affect body energy composition. In fact, despite the similar metabolisable energy intake during the whole 8-week period, fructose-fed rats exhibited higher body energy and the increase in body energy was almost exclusively as lipids. Interestingly, a parallel increase in circulating NEFA and skeletal muscle TAG and ceramide contents was found in fructose-fed rats. It should be noted that the percentage increase in plasma NEFA and muscle ceramide is similar, in agreement with previous results in obese human subjects of a significant correlation between muscle ceramide and plasma NEFA levels(Reference Adams, Pratipanawatr and Berria28).
Rats fed the fructose-rich diet also displayed reduced whole-body glucose tolerance, with a higher plasma insulinaemic response to a glucose load, results that are similar to those previously obtained in rats fed a high-fat diet(Reference Crescenzo, Bianco and Falcone29). Since glucose buffering capacity after a glucose load is primarily determined by the skeletal muscle metabolic response to insulin, downstream target of the insulin signalling pathway in the skeletal muscle of fructose-fed rats was investigated, and no difference was found in p-Akt kinase levels, but when the p-Akt kinase levels were normalised to plasma insulin concentrations, significantly lower values were found in fructose-fed rats compared with the controls. This result is in agreement with increased skeletal muscle ceramide content, since it is well known that ceramide has been identified as a key mediator of insulin resistance via inhibition of Akt phosphorylation(Reference Coen and Goodpaster30), and indicates that insulin action is blunted in skeletal muscle from fructose-fed rats. However, this impairment is compensated by higher plasma insulin, so that the final response of skeletal muscle cells to insulin is maintained. In agreement with this finding, tissue glycogen content does not vary in fructose-fed rats compared with the controls, in line with the well-known positive regulatory role of insulin in glycogen synthesis in this tissue(Reference Yeaman, Armstrong and Bonavaud31).
Whole-body insulin resistance is usually associated with alterations in the metabolic activity of skeletal muscle, and derangement in mitochondrial performance in skeletal muscle has been correlated with the development of insulin resistance(Reference Pagel-Langenickel, Bao and Pang32). Other studies have indicated that insulin resistance can be dissociated from mitochondrial respiratory capacity. Thus, in rat studies where the animals were given a high-fat diet, insulin resistance develops along with an increase in mitochondrial content and an increase in the capacity to oxidise fat(Reference Turner, Bruce and Beale33–Reference Han, Hancock and Jung36). Mitochondrial content inside the cell and oxidative phosphorylation efficiency are the main determinants of mitochondrial performance. Oxidative phosphorylation efficiency depends on the degree of coupling between oxygen consumption and ATP synthesis, which is always lower than 1 and can vary according to the metabolic needs of the cell(Reference Johannsen and Ravussin11). It is well known that fatty acids can act as natural uncouplers of oxidative phosphorylation(Reference Rial, Rodríguez-Sánchez and Gallardo-Vara37). Therefore, mitochondrial mass and the degree of coupling, as well as the uncoupling effect of the fatty acid palmitate were assessed. The present results show that in fructose-fed rats, skeletal muscle mitochondrial mass increases, in agreement with the increased mitochondrial protein synthesis rate found in rats fed a high-fructose diet(Reference Chanseaume, Giraudet and Gryson38). This increase in mitochondrial mass could be a compensatory mechanism to the increased fatty acid supply due to higher plasma NEFA levels found here, since it has been shown that raising plasma NEFA levels induces increased mitochondrial biogenesis in skeletal muscle(Reference Garcia-Roves, Huss and Han39). However, the compensatory increase in mitochondrial mass probably failed to buffer NEFA oversupply, due to the increased mitochondrial coupling found in rats fed a fructose-rich diet, since in this condition, less amount of fuels are oxidised to obtain the same amount of ATP. Decreased substrate burning by skeletal muscle is supported by the decrease in RMR found here in fructose-fed rats compared with the controls, given that skeletal muscle accounts for about 30 % of whole-body energy expenditure(Reference Rolfe and Brown40). The failure of the mitochondrial compartment to face with the increased supply of lipid substrates in fructose-fed rats is also in agreement with their significantly higher skeletal muscle TAG, which imply an increase in intramuscular adipose tissue deposition. Intramuscular adipose tissue enlargement is considered among the determinants of insulin resistance(Reference Vettor, Milan and Franzin41), through diacylglycerol and ceramide accumulation inside the skeletal muscle cells(Reference Martins, Nachbar and Gorjao42).
Another unwanted consequence of the increased degree of coupling is higher mitochondrial free radical production (reactive oxygen species). In fact, the production of reactive oxygen species by the mitochondrial respiratory chain is higher when the membrane potential increases(Reference Korshunov, Skulachev and Starkov43, Reference Mailloux and Harper44), and reactive oxygen species production is indicated as one of the potential causes leading to insulin resistance(Reference Houstis, Rosen and Lander45, Reference Rains and Jain46). For these reasons, the oxidative status of skeletal muscle mitochondria was also assessed and, in fructose-fed rats, signs of oxidative damage were found, together with the decreased activity of SOD, one of the enzymatic components of the antioxidant defence system. These results are in agreement with the impairment in insulin action found in this tissue, since it has recently been shown that the overexpression of mitochondrial SOD in skeletal muscle from rats fed a high-fat diet ameliorated the reduction in muscle glucose uptake and that this effect was mediated through an altered redox state(Reference Boden, Brandon and Tid-Ang47).
In conclusion, the present results give evidence that a fructose-rich diet has a deep impact not only on liver tissue, which is responsible for about 90 % of fructose metabolism, but also on another metabolically relevant tissue, the skeletal muscle. In this tissue, the consequences of high fructose feeding are decreased insulin signalling, stimulated mitochondrial biogenesis and increased mitochondrial coupling. This latter modification could have a detrimental metabolic effect by causing energy sparing that contributes to the high metabolic efficiency of fructose-fed rats.
Acknowledgements
The present study was supported by a grant from the University ‘Federico II’ of Naples and by P.O.R. Campania FSE 2007-13, Project CREME. The authors thank Dr Emilia De Santis for the skilful management of the animal house. S. I. designed and supervised the study. S. I. and G. L. obtained funding and provided administrative, technical and material support. R. C., F. B., P. C., A. M. and L. C. performed the animal experiments. R. C. and S. I. contributed to the analysis of data and the interpretation of the results. R. C., S. I. and G. L. wrote the draft of the manuscript. All the authors critically reviewed the manuscript. The authors declare that there is no conflict of interest.