


The retinal pigment epithelium (RPE), a monolayer of pigmented cells strategically situated behind the photoreceptor cells(Reference Sparrrow, Hicks and Hamel1), plays an important role in maintaining the structural integrity and normal physiological metabolism of the retina as a constituting unit, and in the visual cycle as a functional unit. RPE cells support the survival and normal functioning of photoreceptors by acting as part of the outer blood–retinal barrier (BRB) to control the exchange of nutrients and waste products between underlying choroidal blood vessels and overlying photoreceptors(Reference McBlee, Palczewski and Baehr2, Reference Qin and Rodrigues3). RPE cells also participate in converting and storing retinoid, absorbing scattered light and producing the trophic factors necessary for photoreceptor survival and the immunological factors necessary for establishing the immune privileges of the eye(Reference Sparrrow, Hicks and Hamel1, Reference Qin and Rodrigues3). Considerable evidence supports the idea that RPE injury or dysfunction is a primary cause of retinal pathologies, including monogenic retinal dystrophies, age-related macular degeneration (AMD) and retinal detachment(Reference Zhu, Deng and Xu4, Reference Rattner, Toulabi and Williams5). Ageing is one of the factors that cause the destruction of RPE cells and cellular senescence is considered to manifest the critical ageing process of organisms. Such arguments are supported by in vivo observations of various species, using distinct markers, such as 20S proteasome, p53 and β-galactosidase; in these observations, the number of senescent cells in the tissues was found to increase proportionally with age(Reference Herbig, Ferreira and Condel6–Reference Ressler, Bartkova and Niederegger8).
Senescent cells increase in response to a number of stimuli. Telomere attrition and oncogene activation are the best-understood triggers for cell ageing. Therefore, as an in vitro senescent model, replicative ageing is widely used for its enhanced senescent phenotypes, including shortened telomeres, reduced cell viability, enhanced β-galactosidase activity and ageing morphological characteristics(Reference Foreman and Tang9, Reference Li, Wang and Shen10). A variety of stresses, diseases or pathological conditions, as well as environmental and nutritional factors, also appear to play important roles in cell senescence. However, such senescence is generally telomere-independent(Reference Toussaint, Medrano and Von Zglinicki11).
Clinical and in vitro observations of RPE from persons aged over 60 years indicate that the replicative capacity and functions of cells, such as those of the BRB, are compromised during ageing(Reference Roider, Brinkmann and Wirbelauer12, Reference Rastmanesh13). In ophthalmology, the ageing of RPE cells is now being taken seriously because the age-related dysfunction of RPE cells is considered to be a potential pathogenetic marker of the onset of age-related retinopathies such as AMD(Reference Boulton, Róanowska and Wess14–Reference Chen, Liu and Lukas16). Furthermore, RPE cells are susceptible to injury by excessive light exposure because their surroundings include intense light, usually at wavelengths between 400 and 760 nm, high oxidative stress and high level of PUFA(Reference Algvere, Marshall and Seregard17, Reference Youn, Chou and Cullen18). Given their escalating dependence on computers, an increasing number of people are beginning to suffer eye problems, such as eyestrain, blurred vision and ocular dryness, called computer vision syndrome, as found by the American Optometric Association(Reference Shiraga, Adamus and Shiraga19). Although the pathology of computer vision syndrome is complex, brightness, a key computer screen feature, is thought to be associated with visual fatigue and is considered to be one of the causes of computer vision syndrome(Reference Skilling, Weaver and Kato20). The human retina is protected from high-energy UV light by the cornea and lens, which absorb UV light below 400 nm, but can be damaged by visible light(Reference Boulton, Rozanowska and Rozanowski21). Blue light (400–500 nm) is believed to be the main region of the visible light spectrum involved in damaging retinal tissues due to its relatively high energy and ability to penetrate through tissues to cells and their organelles(Reference Wenzel, Grimm and Samardzija22, Reference Roehlecke, Schaller and Knels23). With regard to light-induced damage to RPE cells, chromophores, formed by rhodopsin intermediates in the photoreceptor outer segments, such as the protein A2E, a major component of lipofuscin, have been considered to be the major source of singlet oxygen in RPE cells(Reference Roehlecke, Schaller and Knels23). Singlet oxygen reacts with A2E to generate epoxides and subsequently disrupt DNA(Reference Sparrow, Vollmer-Snarr and Zhou24). Recent studies have shown that blue light can also damage lipofuscin-free RPE cells by directly inducing the production of reactive oxygen species (ROS) in RPE mitochondria, thus leading to apoptosis(Reference King, Gottlieb and Brooks25, Reference Rezai, Gasyna and Seagle26).
Towards the common goal of determining preventative strategies and effective therapies for eye diseases, food scientists and nutritionists are searching for a diet therapy that will prevent eye diseases through the intake of natural food sources and nutritional supplements. Anthocyanins are reported to be important nutrients capable of protecting or improving one's vision. The possible mechanisms of action of this compound on the visual organ include accelerated resynthesis of rhodopsin(Reference Zheng, Liang and Hao27–Reference Chen, Wu and Dentchev30), modulation of retinal enzymatic activity(Reference Chen, Wu and Dentchev30, Reference Kalt, Hanneken and Milbury31), protection of retinal cells by antioxidation(Reference Sparrow, Vollmer-Snarr and Zhou24, Reference Jang, Zhou and Nakanishi32) and improved microcirculation(Reference Chen, Wu and Dentchev30). Anthocyanins can pass the BRB and be transported to RPE cells, as observed in vivo (Reference Matsumoto, Nakamura and Iido33, Reference Kalt, Berg and Mcdonald34). However, to date, reports on the protective effects of anthocyanins on RPE cells are rare. The aim of the present study is to determine the effects of senescence and light-induced cellular stress on RPE cell health, as well as elucidate the potential ameliorative effects of blueberry anthocyanins in an in vitro culture system.
Materials and methods
Plant materials
Fresh blueberries (10 g; Vacciniun spp.), grown in the Greater Hinggan Mountains in northeast China and mature by late July, were supplied by the Science and Technology Bureau of Greater Hinggan Mountains District in mid-August, 2010. The berries were stored at − 20°C until use.
Chemicals and reagents
An Amberlite XAD-7 used for purifying blueberry polyphenols was obtained from Sigma (Sydney, Australia). A Sephadex LH20 and Oasis HLB cartridge used for isolating and purifying anthocyanins were purchased from Amersham Biosciences AB (Uppsala, Sweden) and Waters (Milford, MA, USA), respectively. Deionised water was produced using a Milli-Q water purification system (Millipore, Billerica, MA, USA). Analytical reagent-grade solvents were used for extraction.
Anthocyanin extraction and purification
Polyphenol separation and anthocyanin purification from blueberries were performed as previously described with several modifications(Reference Kim, Heo and Kim35, Reference Yi, Fischer and Krewer36). Fresh blueberries (50 g) and 100 ml absolute methanol were mixed in a 250 ml round-bottomed flask, and then homogenised using a homogeniser (XHF-D, Ningbo Science & Biotechnology Company, Ningbo, Zhejiang, China). The homogenised sample was further centrifuged for 10 min at 4000 g, and the supernatant was filtered through a moderate speed 102 qualitative filter paper (Hangzhou Special Paper Industry Company Limited, Hangzhou, Zhejiang, China). This procedure was repeated to re-extract the residue. The two filtrates were combined and evaporated using a rotary evaporator at 40°C and a vacuum pressure of 0·1 MPa. Part of the concentrated solution was loaded onto an Amberlite XAD-7 column, and the remaining part was lyophilised (crude extract) for further analysis. After 1 h, the XAD was washed with about 800 ml 1 % (v/v) formic acid aqueous solution to remove non-polyphenolic compounds, after which the polyphenolics were eluted with about 600 ml absolute methanol with 1 % (v/v) formic acid. The eluent was concentrated at 40°C and lyophilised in vacuum using a freeze-dryer (Four-ring Science Instrument Plant Beijing Company Limited, Beijing, China). Then, 48 h later, a friable dark-red powder was obtained. A 50 mg lyophilised sample (polyphenol mixture) was resolubilised in pH 7 phosphate buffer and applied to a Sephadex LH20 column. The column was first washed with a pH 7 phosphate buffer to remove phenolic acids, and then with 70 % (v/v) methanol acidified with 10 % (v/v) formic acid to elute anthocyanins and flavonols. The phenolic acids removed, also called the phenolic acid-rich fraction, were collected and lyophilised for later use. The anthocyanin and flavonol fraction was freeze-dried, resolubilised in 5 % (v/v) formic acid in water, and then applied to an Oasis HLB cartridge. The cartridge was washed with 5 % (v/v) formic acid, followed by ethyl acetate, and then with 10 % (v/v) formic acid in methanol. The ethyl acetate eluted flavonols, and this fraction was called the flavonol-rich fraction after drying in a vacuum at 40°C using a DZF-6021 vacuum drying oven (Hangzhou Lihui Environmental Testing Equipment Company Limited, Hangzhou, China). The anthocyanins were eluted with acidified methanol, and the eluents were freeze-dried and called blueberry anthocyanin extracts (BAE). These extracts or fractions were kept at − 20°C until use.
Determination of total phenolics
The total phenolic content was determined using the Folin–Ciocalteu assay as described by Singleton & Rossi(Reference Singleton and Rossi37), with some modifications. Briefly, 1 ml of diluted samples (100 μg/ml) was mixed with 0·5 ml of 0·2 mol/l Folin–Ciocalteu reagent in 10 ml volumetric flasks. After 5 min, 1·5 ml of a 20 % sodium carbonate solution was added. The volume was then increased to 10 ml with distilled water and mixed thoroughly. After 30 min at room temperature, the absorbance was measured at 760 nm using a spectrophotometer (722S, Shanghai Precision & Science Instrument Company Limited, Shanghai, China), and the total phenolic content was calculated using the standard curve of gallic acid. Results were expressed as weight percentages, which were calculated as follows: gallic acid equivalent (GAE)/wet weight of extracts or fractions × 100.
Determination of total anthocyanins
Total anthocyanin content was measured using a pH differential method described previously(Reference You, Wang and Chen38, Reference Giusti, Wrolstad and Wrolstad39). Two dilutions of the extracts were prepared, one for pH 1·0 using potassium chloride buffer and the other for pH 4·5 using sodium acetate buffer. The samples were diluted to a final concentration of 50 μg/ml. The absorbance was then measured at 515 nm with distilled water as a blank. The samples showed no haze or sediment; thus, correction at 700 nm was omitted. The total anthocyanin content was then determined using Lambert–Beer's law, which was calculated as follows:
where A is the difference in the absorbance at 515 nm between pH 1·0 and 4·5, MW is 449·2 (the molecular weight of cyanidin-3-glucoside (g/mol)), DF represents a dilution factor and ɛ denotes the extinction coefficient of cyanidin-3-glucoside (26 900 L × mol− 1 × cm− 1, where L (path length) = 1 cm). Results were expressed as weight percentages, which were calculated as follows: (cyanidin-3-glucoside equivalent/wet weight of extracts or fractions) × 100.
Cell culture
A human RPE cell line, ARPE-19 (ATCC CRL-2302; American Type Culture Collection, Manassas, VA, USA), was used in the present study and cultured as previously described(Reference Hui, Yi and Fengling40). Cell cultures were grown in Dulbecco's modified Eagle's/Ham's F12 media (Invitrogen, Carlsbad, CA, USA) supplemented with 10 % fetal bovine serum (Sigma-Aldrich, St Louis, MO, USA), containing 1 % antibiotic mixture of penicillin (100 U/ml) and streptomycin (100 mg/ml; Invitrogen) at 37°C under a humidified 5 % CO2 atmosphere.
Blueberry anthocyanin extract cytotoxicity
Cell viability was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT; Sigma-Aldrich) assay as previously described(Reference Zhang and Popovich41). RPE cells were seeded in ninety-six-well plates (Corning–Costar, Corning, NY, USA) at a concentration of 5 × 105 cells/ml, and then allowed to attach after 48 h. The medium was then replaced with serum-free F12 medium containing 1·0 × 10− 3, 1·0 × 10− 2, 0·1, 1·0, 10·0 and 100·0 μg/ml BAE, respectively. Then, 24 h later, the cell supernatant was removed for lactate dehydrogenase (LDH) assay. About 200 μl of 0·5 mg/ml MTT serum-free F12 medium were added into each well of the plates, after which incubation was performed for 4 h. After removal of the MTT solution, 150 μl of dimethyl sulfoxide (Sigma-Aldrich) were added, and the absorbance was measured at 570 nm using a plate reader (Molecular Devices Company, Sunnyvale, CA, USA). Results were expressed as the percentage of viable cells with respect to untreated control cells. Cell viability (%) was calculated as follows: ((mean absorbance of the sample − reference absorbance)/mean absorbance of the control) × 100. The cellular release of LDH following BAE exposure was used as a measure of cellular damage/integrity. Enzymatic activity was determined using an LDH kit (Beyotime Institute of Biotechnology, Jiangsu, China) according to the manufacturer's instructions.
Replicative senescent model
Cell proliferation activity
The fourth passage of RPE cells was subcultured at 80–90 % confluence with a seeding density of 2 × 105/ml. Each subculture was deemed as one passage. The control group was cultured according to the methods described previously. To evaluate the anti-ageing activity of BAE on RPE cells, extra BAE was added to the culture medium at a final concentration of 0·1 μg/ml. In each subculture, the MTT assay was used to assess the proliferative capability of RPE cells.
Senescence-associated β-galactosidase activity
Senescence was investigated using a senescence-associated β-galactosidase staining kit (Beyotime Institute of Biotechnology) according to the manufacturer's instructions. The treated RPE cells were washed twice with pH 6·0 PBS, and then fixed with 2 % formaldehyde and 0·2 % glutaraldehyde in PBS at room temperature for 4 min. Cells were then washed twice with PBS and incubated in the dark for 8 h at 37°C with fresh β-galactosidase staining solution (1 mg/ml 5-bromo-4-chloro-3-indoyl-β-d-galactopyranoside, 40 mm-citric acid/sodium phosphate, pH 6·0, 5 mm-potassium ferrocyanide, 5 mm-potassium ferricyanide, 150 mm-NaCl and 2 mm-MgCl2 diluted in PBS). Cells were then examined to determine the development of blue colouring and photographed using a light microscope (Chongqing Optical and Electrical Instrument Company Limited, Chongqing, China). The percentage of blue-stained cells (cells of active β-galactosidase) was calculated as the number of blue-stained cells/the number of total cells. For analysis, five visual fields of each passage group were chosen, and each field contained at least 150 cells. The results were recorded as means and standard deviations of the five counts.
Light-induced damage model
Light exposure
The RPE cells were subjected to white light irradiation by an integrated light-emitting diode lamp system designed by the authors. The light-emitting diode lamps were purchased from Foshan Nationstar Optoelectronics Company Limited (Foshan, Guangdong, China), and the spectrum of the light was 420–800 nm according to the introduction provided by the manufacturer. The light-emitting diode lamps were set into a twenty-four-well plate and assembled together with wires, switches and knobs. Light intensity was adjusted through the knobs and measured with a TES-1330A light meter (TES Electrical Electronic Corporation, Taipei, Taiwan). RPE cells in an active growing state were planted into plates with serum-containing F12 medium at a concentration of 5 × 105 cells/ml. After 85 % of the cells were adhered to the wall, the medium was replaced with serum-free F12 medium for both the negative (no light exposure, no BAE treatment) and the positive (light exposure, no BAE treatment) controls. For the BAE treatment groups (light exposure, BAE treatment), serum-free F12 medium containing 0·1, 1·0 or 10·0 μg/ml of BAE was added. The RPE cells were then exposed to 2500 (sd 500) lx white light (420–800 nm) for 12 h. In addition, black adhesive tape was placed on the cover of the plates in the control group to avoid exposure to light. All these procedures are summarised in Fig. 1.
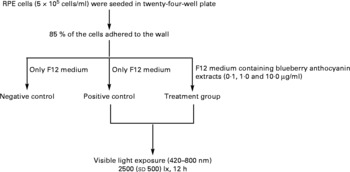
Fig. 1 Scheme of light exposure. After 85 % of the retinal pigment epithelium (RPE) cells adhered to the walls of the twenty-four-well plate, the supernatants were replaced by serum-free F12 medium with or without blueberry anthocyanin extracts (BAE). Then, RPE cells were irradiated with visible light (420–800 nm) at 2500 (sd 500) lx for 12 h. Negative control: no light exposure, no BAE treatment; positive control: light exposure, no BAE treatment; treatment group: light exposure, BAE treatment.
Cell viability assay
After subjecting to light irradiation, the culture medium was replaced with serum-containing F12 medium. RPE cells were continuously incubated in this medium for 24 h under the same normal conditions described previously, before cell viability was finally analysed using an MTT assay.
Vascular endothelial growth factor detection
The amount of vascular endothelial growth factor (VEGF) in the supernatant of the RPE culture was determined by ELISA. After light irradiation, the culture media were collected and then centrifuged at 1000 g for 5 min. The VEGF level in the culture medium was analysed by a commercial VEGF ELISA kit (Shanghai ExCell Biology, Inc., Shanghai, China) according to the manufacturer's instructions. Absorbance at 450 nm was measured using a plate reader (Molecular Devices Company).
Cellular reactive oxygen species measurement
The ROS level in RPE cells was monitored using the fluorescent dye 2,7-dichlorodihydro-fluorescein diacetate (DCFH-DA; Sigma-Aldrich). The membrane-permeable DCFH-DA enters the cells and produces a fluorescent signal after intracellular oxidation by ROS (such as H2O2 and lipid peroxides), and the fluorescence can directly reflect the overall intracellular ROS(Reference Wilhelm, Vytasek and Ostadalova42). RPE cells were cultured on the bottom of a transparent black ninety-six-well plate (Corning–Costar) in 200 μl of the growth medium at a density of 5 × 105 cells/ml. After 48 h incubation, the medium was replaced with serum-free F12 medium containing 0·1, 1·0 or 10·0 μg/ml of BAE. For the control group, no BAE was present in the medium. The RPE cells were then exposed to 2500 (sd 500) lx visible light for 12 h. Then, the new medium with 25 μm-DCFH-DA was added into the ninety-six-well plate, and the cells were incubated sequentially for another 1 h at 37°C. After the supernatant containing DCFH-DA was removed, the cells were carefully washed twice with Hanks’ balanced salt solution (Gibco Life Technologies, Grand Island, NY, USA). The fluorescence of the cells from each well was recorded by a multifunctional microplate reader at 530 nm emission and 485 nm excitation. Subsequently, the protein content of each well was determined using a commercial kit (Beyotime Institute of Biotechnology), and the ROS level of each well was calculated as the fluorescence intensity of each well/the protein content of each well.
Statistical analysis
The statistical significance of the differences between the control and treatment groups was analysed by one-way ANOVA using Origin version 8.0, followed by Tukey tests. A normality test showed that all the raw data had a normal distribution, and all groups were determined to have equal variance by a variance test. Data were expressed as the means and standard deviations of at least three individual experiments, each run in triplicate. P < 0·05 (two-sided) was considered significant.
Results and discussion
Extraction and fractionation
To verify whether or not the anthocyanin-rich fraction was clearly separated through the processes, the total polyphenol and total anthocyanin contents of the different extracts or fractions were analysed, and the results are shown in Table 1. After the first-stage of purification with Amberlite XAD-7, the total polyphenol and total anthocyanin concentrations in the polyphenol mixture improved markedly, with both contents nearly fourteen times higher than those in the crude extracts. After further purification, the polyphenol mixture was fractionated into anthocyanin-rich, phenolic acid-rich and flavonol-rich fractions, and their weight yields were approximately 40·2, 38·3 and 4·5 %, respectively. Anthocyanins and phenolic acids are evidently the main components in blueberry polyphenols. The flavonol-rich fraction had relatively high total polyphenol contents (approximately 88 %) compared with the other two fractions. It is believed that flavonol compounds are another main constituent of blueberry polyphenols. The highest purity of anthocyanins was observed in the anthocyanin-rich extracts (nearly 31 %), and only small amounts of anthocyanins were detectable in the other two fractions. These results demonstrate that the anthocyanins are purified effectively from blueberries via the method employed.
Table 1 Weight yield, total phenol content and total anthocyanin content of different blueberry extracts/fractions*
(Mean values and standard deviations)

GAE, gallic acid equivalents; CGE, cyanidin-3-glucoside equivalents.
* All experiments were performed in triplicate and values are averages of triplicate analyses and standard deviations.
† Weight yield of each fraction was calculated as each fraction weight/polyphenol mixture weight × 100 %.
‡ Total polyphenols are expressed as GAE, and the total polyphenolic contents percentage was calculated as GAE/fraction weight × 100 %.
§ Total anthocyanins are expressed as CGE, and the total anthocyanin contents percentage was calculated as CGE/extract weight × 100 %.
The measured total anthocyanin content in the anthocyanin-rich extracts was apparently lower than the expected value, which should be more than 50 %. This condition may be associated with the pH differential method employed in the present study. According to previous reports, the total anthocyanin content obtained by HPLC was 2·0–2·3 times higher than the values from the pH differential method(Reference Lee and Finn43). In addition, the total anthocyanin content expressed as malvidin-3-glucoside is much higher than the content expressed as cyanidin-3-glucoside(Reference Lee, Rennaker and Wrolstad44). Therefore, analysis suggests that the real content of total anthocyanins in the anthocyanin-rich extracts could be significantly more than 31 %.
Cytotoxicity of blueberry anthocyanin extracts on retinal pigment epithelium cells
The dose–response cytotoxicity of BAE on RPE cells is shown in Fig. 2. Compared with the control cells, no significant BAE toxicity effects on RPE cells occurred at concentrations ranging from 1·0 × 10− 3 to 10 μg/ml over a 24 h period. BAE at a concentration of 0·1 μg/ml could improve RPE cell proliferation to approximately 1·25-fold that of the control (P < 0·05; Fig. 2(a)). Compared with the control, LDH release was not significant when the concentrations of BAE were below 100 μg/ml (Fig. 2(b)). At 100 μg/ml, the cytotoxicity of BAE on RPE cells appeared, as indicated by the remarkable increase in LDH release. Thus, a dose of 0·1 μg/ml of BAE was selected for the replicative ageing model, and doses for light-induced damage ranged from 0·1 to 10 μg/ml.

Fig. 2 Cytotoxic effect of blueberry anthocyanin extracts (BAE) on retinal pigment epithelium (RPE) cells. After RPE cells (5 × 105 cells/ml) were incubated with Dulbecco's modified Eagle's/Ham's F12 medium containing 10 % serum for 48 h, the cells were exposed to serum-free F12 medium containing 1·0 × 10− 3 to 100·0 μg/ml of BAE for 24 h. (a) Cell viability was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay and is expressed as a percentage of control cells with BAE-free F12 medium. (b) Cytosolic lactate dehydrogenase (LDH) activity was also evaluated. Enzyme activity is expressed as a ratio of the activity per g protein. Values are means, with standard deviations represented by vertical bars (n 3). * Mean values were significantly different from those of the control group (P < 0·05; one-way ANOVA).
Depressant effect on senescence of retinal pigment epithelium cells
The senescence associated with RPE changes can lead to an abnormality in its physiological function and is closely associated to the development of AMD(Reference Feher, Kovacs and Artico45). The possible mechanisms of cellular senescence include telomere shortening and oxidative stress(Reference Toussaint, Medrano and Von Zglinicki11). Unfortunately, no in vitro cell model can completely reflect the complicated conditions of cell ageing. In the present work, the effects of subculture and light exposure on senescent characteristics of RPE cells were investigated.
The positive stain of β-galactosidase clearly increased significantly with cell passage, as found during morphologic observations with an optical microscope (Fig. 3(a1–f1)), whereas an obviously slowing trend was observed when the RPE cells were treated with 0·1 μg/ml of BAE (Fig. 3(a2–f2)). The same phenomenon was also observed in the RPE cell proliferation according to the MTT assay. Compared with the fifth passage of RPE cells, the number of RPE cells decreased sharply starting from the seventh passage, and approximately 14·6 % of the viable cells remained at the tenth passage (Fig. 3(g)). Meanwhile, from the fifth to the tenth passage, the percentage of β-galactosidase positive cells increased sharply from 30·1 (sd 2·5) to 92·5 (sd 8·2) %, respectively. In contrast, 76·9 (sd 3·6) % of the cells remained alive at the tenth passage, and the percentage of senescent cells decreased from 22·5 (sd 1·7) to 58·2 (sd 5·1) % (Fig. 3(h)) when the RPE cells were treated with 0·1 μg/ml of BAE.

Fig. 3 Characterisation of replicative ageing of retinal pigment epithelium (RPE) cells in culture and the antidotal activity of blueberry anthocyanin extracts (BAE). Cells from the fifth to the tenth passage were employed. RPE cells without BAE treatment were defined as control (![]() ); the inhibitory effect of BAE on the ageing of RPE cells was evaluated at a dose of 0·1 μg/ml (
); the inhibitory effect of BAE on the ageing of RPE cells was evaluated at a dose of 0·1 μg/ml (![]() ). The cell morphology was observed by a light microscope ( × 100), and positive senescent cells in each group are displayed in blue (
). The cell morphology was observed by a light microscope ( × 100), and positive senescent cells in each group are displayed in blue (![]() ). (a1–f1) Natural ageing process in different passages of RPE cells, and (a2–f2) demonstrate the corresponding passages treated with BAE. (g) The cell viability of each passage, expressed as optical density. (h) Senescent percentage of each group. Values are means, with standard deviations represented by vertical bars (n 3). ** Statistical comparisons between BAE-treated groups and controls were carried out in cells of the same passage, and mean values were significantly different (P < 0·01; one-way ANOVA).
). (a1–f1) Natural ageing process in different passages of RPE cells, and (a2–f2) demonstrate the corresponding passages treated with BAE. (g) The cell viability of each passage, expressed as optical density. (h) Senescent percentage of each group. Values are means, with standard deviations represented by vertical bars (n 3). ** Statistical comparisons between BAE-treated groups and controls were carried out in cells of the same passage, and mean values were significantly different (P < 0·01; one-way ANOVA).
The results also showed that after light exposure at 2500 (sd 500) lx for 12 h, RPE cells in the fourth passage exhibited premature senescence (Fig. 4). Compared with the negative control (33·3 (sd 2·9) % of senescent cells), the percentage of senescent cells in the positive control increased to 57·2 (sd 6·0) %. Besides, the ROS level in RPE cells was also significantly (P < 0·01) higher than that in the negative control (Fig. 5). When RPE cells were treated with 1·0 or 10·0 μg/ml of BAE, the percentage of senescent cells decreased to 35·1 (sd 4·3) and 32·5 (sd 5·3) %, respectively, and the intracellular ROS levels in both treatments decreased to approximately 21 %. A concentration of 0·1 μg/ml of BAE was not efficient in suppressing cell ageing and intracellular ROS level induced by light. These results indicate that the protective effect of BAE on light-induced ageing of RPE cells may be associated with the pathway of suppressing oxidative stress.

Fig. 4 Protective effect of blueberry anthocyanin extracts (BAE) on visible light-induced (420–800 nm) senescence of retinal pigment epithelium (RPE) cells. Confluent cultures were exposed to light (2500 (sd 500) lx) or maintained in the dark for 12 h. (a) Morphology of RPE cells as observed by a light microscope ( × 100) after β-galactosidase staining; senescent cells are displayed in blue (![]() ). (b) Senescent percentage of RPE cells in the negative control, positive control and treatment groups. Negative control: no light exposure, no BAE treatment; positive control: light exposure, no BAE treatment; treatment group: light exposure, BAE treatment. Values are means, with standard deviations represented by vertical bars (n 3). ** Mean values were significantly different (P < 0·01; one-way ANOVA).
). (b) Senescent percentage of RPE cells in the negative control, positive control and treatment groups. Negative control: no light exposure, no BAE treatment; positive control: light exposure, no BAE treatment; treatment group: light exposure, BAE treatment. Values are means, with standard deviations represented by vertical bars (n 3). ** Mean values were significantly different (P < 0·01; one-way ANOVA).

Fig. 5 Effect of blueberry anthocyanin extracts (BAE) on visible light-induced (420–800 nm) increases in intracellular reactive oxygen species (ROS). Confluent cultures were exposed to light (2500 (sd 500) lx) or maintained in the dark for 12 h. The amounts of intracellular ROS were monitored using 2,7-dichlorodihydro-fluorescein diacetate. Negative control: no light exposure, no BAE treatment; positive control: light exposure, no BAE treatment. Values are means, with standard deviations represented by vertical bars (n 3). ** Mean values were significantly different (P < 0·01; one-way ANOVA).
Li et al. (Reference Li, Wang and Shen10) reported that the viability of RPE cells is reduced significantly with the number of passages and that the percentage of β-galactosidase-positive cells increased from 8·31 (sd 1·41) to 45·86 (sd 2·42) % from the second to the twenty-fourth passage, respectively(Reference Li, Wang and Shen10). In vivo studies have found an age-associated increase in the occurrence of senescent cells in normal tissues(Reference Jeyapalan and Sedivy46). More than 15 % of the dermal fibroblasts in very old baboons showed senescent phenotypes, as determined by the presence of damaged telomeres(Reference Jeyapalan and Sedivy46). Under the conditions employed, the results of the present study further confirm the close relationship between cell apoptosis and senescence. It is believed that the ageing process is accompanied by apoptosis, as indicated by the common genes expressed in both apoptosis and senescence(Reference Bree, Stenson-Cox and Grealy47, Reference Seluanov, Gorbunova and Falcovitz48). With regard to the mechanisms of replicative ageing, telomere shortening has been considered to be the major mechanism to which the replicative ageing of post-mitotic cells, including RPE cells(Reference Passos and Zglinicki49), may be attributed. In recent studies, the mammalian target of rapamycin-mediated pathways, one of the central players in regulating ageing, manifested age-associated changes in human RPE cells in a replicative senescence model(Reference Chen, Wang and Cai50).
Stress conditions, which are generally telomere-independent, can likewise induce senescence of RPE cells. This phenomenon may be due to the susceptibility of RPE cells to suffering from oxidative stress in vivo from their physiological environment in the retina, which includes high contents of PUFA, high levels of aggregated light and high levels of oxygen stress. In the present study, visible light exposure was shown to increase intracellular ROS levels and accelerate RPE cell ageing. Previous reports have also shown that, after repeated near-UV irradiation, RPE proliferation is markedly suppressed and significantly exhibits the senescent characteristics of RPE cells through down-regulation of pigment epithelium-derived factor and tissue inhibitor of metalloproteinase 3 gene expression, both of which are used as markers of senescence(Reference Li, Yanoff and Li51). Moreover, in vitro studies have shown that exposure to mild hyperoxia or chemical oxidants, such as H2O2 and tert-butylhydroperoxide, could also induce cellular senescence in human RPE cells(Reference Honda, Hjelmeland and Handa52–Reference Kook, Wolf and Yu54), the pathway for which may be associated with the release of a transforming growth factor-α(Reference Yu, Fuchahofer and Kook53) and the accumulation of large fragments of cytosolic proteins(Reference Kook, Wolf and Yu54). Both LDL and oxidised-LDL can increase transforming growth factor-α expression in human RPE cells. The latter can also accelerate dose-dependency at the onset of RPE senescence, thus providing further information on the effects of LDL and oxidised LDL in human RPE and their potential role in the pathogenesis of AMD(Reference Yu, Lorenz and Haritoglou55).
Antioxidants have previously been demonstrated to decelerate RPE cell ageing. Quercetin, a natural plant-derived antioxidant has been reported to show protective effects on RPE cells with oxidative damage and cellular senescence in vitro (Reference Welge-Lussen56). Melatonin, a well-known antioxidant naturally present in the eyes at night, exhibits potential anti-ageing benefits through down-regulation of human telomerase RT and stimulation of telomerase activity in RPE cells(Reference Rastmanesh13). In the present study, BAE suppressed the ageing of RPE cells by extending their lifespan, reducing the number of positive senescent cells after subculturing and inhibiting increases in intracellular ROS after visible light exposure. After RPE cells were passaged to the tenth generation and treated with 0·1 μg/ml BAE, live cells increased to approximately five times their original values (from 14·6 to 76·9 %), and the percentage of senescent cells decreased by nearly 1·6 times the original number (from 92·5 to about 58·2 %). As well, after visible light exposure, intracellular ROS and the percentage of ageing cells decreased by about approximately 20 and 40 %, respectively, when RPE cells were treated with 1·0 or 10·0 μg/ml BAE.
Protective effect of blueberry anthocyanin extracts on light-induced cell death and vascular endothelial growth factor expression
Light exposure accelerates not only RPE cell ageing, but also cell death and VEGF expression. Fig. 6 shows the protective effect of BAE on RPE cells after light exposure at 2500 (sd 500) lux for 12 h. Cultures in the positive control showed a significant decrease in cell viability (P < 0·01), yet a significant increase in VEGF release (P < 0·01), indicating that light exposure had a direct phototoxic effect on RPE cells. However, the condition was ameliorated after light irradiation where cells were treated with 0·1, 1·0 and 10·0 μg/ml of BAE. The cell viability of RPE cells treated with 1·0 or 10·0 μg/ml BAE was significantly improved compared with the positive control (P < 0·01), especially at a dose of 10·0 μg/ml; at this dose, cell viability exceeded even that of the negative control. Accordingly, after light exposure, VEGF release also significantly (P < 0·01) decreased when RPE cells were treated with 0·1 or 1·0 μg/ml BAE. However, 10·0 μg/ml BAE did not ameliorate VEGF secretion in RPE cells during light exposure. These results indicate that BAE exerts protective effects on RPE cells against light-induced damage.

Fig. 6 Cytoprotective effect of blueberry anthocyanin extracts (BAE) on cell viability and vascular endothelial growth factor (VEGF) secretion of retinal pigment epithelium (RPE) cells after light (420–800 nm) exposure at 2500 (sd 500) lx for 12 h. (a) Effect of BAE on cell viability of RPE cells. (b) Effect of BAE on VEGF secretion in RPE cells. Negative control: no light exposure, no BAE treatment; positive control: light exposure, no BAE treatment. Values are means, with standard deviations represented by vertical bars (n 3). ** Mean values were significantly different (P < 0·01; one-way ANOVA).
The intensity of light is closely associated with the degree of light-induced retinal damage. In normal classrooms and offices, according to the authors’ measurement results, the light intensity is 300–700 lx. Based on the product description of the TES-1330A light meter provided by a TES Electrical Electronic Corporation, the light intensity in most drawing offices, precision testing rooms and design rooms is 1500–3000 lx, while the light intensity in an eye examination room can reach more than 5000 lx. A previous study sought to determine the light intensities of 10 (table lamp) and 36-Watt fluorescent lamps, as well as natural outdoor light. Approximately 30 cm away from the tubes, the light intensity of a table lamp and fluorescent lamp was found to be 1520 (sd 178) and 2260 (sd 343) lx, respectively. On sunny days, from the end of May to the beginning of June in Beijing, the light intensity of natural outdoor light at 09.00, 12.00 and 18.00 hours was 18 512 (sd 1481), over 20 000 and 7824 (sd 239) lx, respectively. However, determination of how much light actually reaches the retina is challenging due to the dual effects of the ocular refractive system (including cornea, aqueous humour, lens and vitreous body) on filtering and focusing light. In this study, a light intensity of approximately 2500 (sd 500) lx was employed to irradiate RPE cells in vitro.
It is believed that, in the entire light spectrum, visible light contributes the largest to the light-induced damages of RPE cells(Reference Youn, Chou and Cullen18, Reference Roehlecke, Schaller and Knels23, Reference Sparrow, Vollmer-Snarr and Zhou24). In the visible spectrum, short wavelengths of light, such as blue light, are believed to be the most hazardous to the retina(Reference Youn, Chou and Cullen18). Excessive exposure to light can lead to the destruction of membrane integrity(Reference Jang, Zhou and Nakanishi32), as well as mitochondrial and nuclear damage(Reference Youn, Chou and Cullen18). These conditions can lead to the degeneration of functions, and even death of RPE cells, which may be executed by caspase-3 and regulated by B-cell lymphoma gene 2(Reference Sparrow and Cai57). According to a previous report, excessive light exposure can result in the over-expression of VEGF in RPE cells, indicating that excessive light exposure is involved in the acceleration of AMD development(Reference Hui, Yi and Fengling40).
In the present study, BAE was shown to be efficient in protecting RPE cells against light-induced damage by suppressing cell death and mediating VEGF release. These possible protective mechanisms may be related to suppressing oxidative damage, ameliorating mitochondrial function and mediating the expression of related enzymes. According to previous reports, bilberry anthocyanin extracts showed the ability to suppress the photo-oxidation of A2E by quenching singlet oxygen, with anthocyanin fractions exhibiting a resistance to membrane permeabilisation when RPE cells were exposed to light at 430 nm(Reference Sparrow, Vollmer-Snarr and Zhou24, Reference Jang, Zhou and Nakanishi32). Mitochondria play a critical role in mediating cell death by expressing associated apoptotic factors when cells are immersed in a stress state(Reference Portt, Norman and Clapp58). When RPE cells were subjected to oxidative stress, bilberry anthocyanin-enriched extracts were found to be able to improve mitochondrial functions and affect the expression of haemeoxygenase-1(Reference Milbury, Graf and Berg59), a stress-inducible cytoprotective enzyme and mediator of VEGF production(Reference Penumathsa, Koneru and Samuel60).
Except for anthocyanins, other polyphenols in blueberries may also be beneficial to eye health. However, no sufficient evidence has yet confirmed that other polyphenolic compounds can be transported into the various regions of the eye. Among known polyphenols or flavonoids, only anthocyanins have been directly studied in animal eyes until recently(Reference Matsumoto, Nakamura and Iido33, Reference Kalt, Berg and Mcdonald34). Not all functional compounds can reach the retina due to the outer BRB, which is structurally and functionally similar to the blood–brain barrier(Reference Mennel, Peter and Meyer61). BRB is established by tight junctions, including the fenestrated endothelium of the choriocapillaris, Bruch's membrane and RPE cells(Reference Mennel, Peter and Meyer61). To avoid confusion with other polyphenols or flavonoids, blueberry anthocyanins were specifically studied in the present experiment. Given that other functional compounds, such as carotenoids (including lutein, zeaxanthin and β-carotene) and vitamins (including vitamins A, C and E), have also been discovered in the retina, their benefits on RPE cells should be considered in future research.
Although anthocyanins have been examined in the eyes, the mechanism by which anthocyanins are metabolised in the eyes remains unclear. According to previous studies(Reference Kalt, Berg and Mcdonald34), rather than rapidly reaching equilibrium with anthocynanins in the blood, anthocyanins accumulate in the eyes after long-term consumption. Malvidin glycosides have been reported to be predominant anthocyanins found in the eyes after blueberry consumption(Reference Matsumoto, Nakamura and Iido33, Reference Kalt, Berg and Mcdonald34), and have been confirmed as the main antioxidant constituents in blueberry anthocyanins(Reference Hurst, Wells and Hurst62). On the basis of the authors’ previous studies, malvidin glycosides were confirmed to be the predominant anthocyanins in blueberries, representing>38 % of the total anthocyanin content of the fruit(Reference Liu, Song and Han63). This result indicates that malvidin glycosides may be the main bioactive anthocyanins that protect RPE cells against ageing and light-induced damage. Earlier studies have mainly focused on the original form of anthocyanins in the eye regions. Kay et al. (Reference Kay, Mazza and Holub64) reported that approximately two-thirds of the anthocyanins consumed present as methylated and glucuronidated metabolites in plasma and urine. Therefore, whether or not other forms of anthocyanins exist in the eyes, should be investigated in future studies.
Based on the present experimental results and those of previous studies, blueberry anthocyanins are confirmed to be effective in preventing RPE cells from senescence and incurring light-induced damage, revealing further that the protective mechanisms of blueberries have visual benefits. The findings indicate that blueberries, or other kinds of fruits that are rich in anthocyanins, have the potential to prevent AMD and other retinal diseases related to RPE cells.
Acknowledgements
The present work was supported by the National Key Technology R&D Program for the twelfth 5-year plan, People's Republic of China (project no. 2011BAD08B03-01). All authors contributed to the design and discussion of the study. Y. L. undertook all the experiments, analysed the data and wrote the manuscript; X. S. performed the study and analysed the data; D. Z., F. Z. and D. W. performed the study; Y. W. analysed the data; F. G., L. X. and G. J. analysed the data and revised the manuscript; and W. W. and B. J. supervised the experiments. All authors approve of the final manuscript and declare no conflicts of interest.







