


Dried blood spotting (DBS) is a minimally invasive technique to obtain blood samples on cards of filter paper for biochemical or genetic analysis. A small prick in a fingertip is enough to produce a drop of blood suitable for sampling. This approach has the advantages of being considerably less invasive than venous blood sampling, does not require healthcare professionals and can be performed by most people on themselves after relatively little training. In addition, if the analyte is stable in dried whole blood, samples can be transported at room temperature (RT) by regular mail. Infrastructure for collecting venous blood samples, separation of plasma and storage and shipment of frozen samples is no longer required. In practice, blood sampling can be carried out anywhere and at any time. This offers significant benefits, especially for nutrient status determination in populations at remote locations or at home( Reference Demirev 1 , Reference McDade, Williams and Snodgrass 2 ).
These advantages make the methodology ideal for use in an internet-based intervention such as the Food4Me study, where all data were self-reported and biological samples were collected remotely by the participants( Reference Celis-Morales, Livingstone and Marsaux 3 ). To date, the Food4Me study is the largest multi-centre, internet-based, personalised nutrition intervention aiming to compare the effectiveness of three levels of personalised nutrition (based on dietary, phenotypic and genotypic data) on behavioural changes (diet and physical activity) and health outcomes (blood metabolites and obesity-related anthropometrics)( Reference Celis-Morales, Livingstone and Marsaux 3 ). The DBS technique was first introduced for diagnosing phenylketonuria in newborns by measuring whole-blood phenylalanine concentration( Reference Guthrie and Susi 4 ), and has since been applied to multiple metabolites( Reference Demirev 1 , Reference Tanna and Lawson 5 ). A critical success factor for assessment of many vitamins and other micronutrients in DBS is their stability on the sampling cards. The compounds need to be stable in the presence of O2 at ambient temperature for at least the initial drying time and shipment time to the laboratory. In the case of the Food4Me study, this period was about 1 week. Stability can be increased by adding reagents such as antioxidants to the sampling cards. However, this approach has limitations due to safety concerns as the impregnated card comes in close contact with the pricked fingertip. To date, DBS assays reported for micronutrients include the vitamins A( Reference Shi, Ma and Humphrey 6 , Reference Craft, Haitema and Brindle 7 ), B12 Reference O’Broin and Kelleher (8) , D( Reference Eyles, Anderson and Ko 9 – Reference Larkin, Gebretsadik and Koestner 13 ), K( Reference Motohara, Endo and Matsuda 14 ) and folate( Reference O’Broin and Gunter 15 ). However, only the assays for vitamins A and D appear to be used frequently. Although vitamin A was reported to be somewhat unstable with a loss of >10 % over the 1st week( Reference Erhardt, Craft and Heinrich 16 ), no stability issues have been reported for vitamin D (25-hydroxy vitamin D3 (25(OH)D3)). An explanation for the particular stability of this vitamin may be the presence of a specific vitamin D-binding protein in blood that (together with serum albumin) binds >99 % of the circulating 25-hydroxy vitamin D( Reference Bikle, Gee and Halloran 17 , Reference Al-oanzi, Tuck and Raj 18 ), thereby stabilising the vitamin on the DBS cards. Vitamin D assays based on DBS have been reported by three independent research groups viz., Eyles et al.( Reference Eyles, Anderson and Ko 9 ) and Newman et al.( Reference Newman, Brandon and Groves 11 ) and by Higashi et al.( Reference Higashi, Suzuki and Hanai 12 ). Although all methods focus on 25(OH)D3, some procedures also include related metabolites, including 25-hydroxy vitamin D2 (25(OH)D2) and 3-epi-25-hydroxy vitamin D3. The main differences between the assays are in the detection and calibration methods used and the extent of validation data that have been published. To date, all methods have been based on reversed-phase liquid chromatography (LC)-MS/MS with electrospray ionisation (ESI+) detection. Eyles and Higashi derivatised the analytes with a N-containing Diels-Alder reagent (4-phenyl-1,2,4-triazoline-3,5-dione), which increases the sensitivity during MS-detection significantly, but has two drawbacks: it introduces an additional time-consuming step during sample preparation, and the derivatisation leads to formation of two stereoisomers that complicate chromatography. The method by Newman et al.( Reference Newman, Brandon and Groves 11 ) does not use analyte derivatisation, but starts with four large punches of filter paper that limit automated handling in small vials during sample preparation. A later publication from the same group indicated that the method has been modified and now also includes derivatisation of the analytes( Reference Larkin, Gebretsadik and Koestner 13 ). However, this approach requires by far the largest sample aliquots, that is, four 6-mm-diameter punches.
In general, calibration of DBS analysis is difficult due to the absence of blank samples in the case of endogenous analytes, and the fact that current reference analytics is performed using plasma or serum samples and not (dried) blood. All existing methods for vitamin D analysis share a quite tedious calibration approach using spiked samples of blood, vitamin D-depleted blood or plasma for calibration.
The aim of the present study was to develop a novel, quicker assay for 25(OH)D3 and 25(OH)D2 from DBS without chemical derivatisation to cope with the expected large sample numbers from the Food4Me study, which might have utility for future large studies and surveys. We focused particularly on improving the calibration methodology to allow direct comparison of the results with published vitamin D status data, which are derived typically from measurements using plasma. Aspects of method development are reported, together with method validation and performance data. The Food4Me study did not include venous blood sampling, which precluded direct comparison of the results obtained from unsupervised collection of DBS by the participants at home with a reference method. Quality measures including spot quality and consistency of status levels of each participant measured at each time point have been assessed. The vitamin D status results were correlated with variables such as seasonality and research centre and were compared with literature data.
Methods
Study design and participants( Reference Celis-Morales, Livingstone and Marsaux 3 )
The Food4Me intervention study was designed as a pan-European randomised controlled trial (RCT) to determine whether providing personalised dietary advice leads to greater improvements in eating patterns and health outcomes compared with a conventional population-based general guidelines approach. Seven research centres in seven European countries participated with more than 220 participants from each centre. DBS samples were collected and analysed at three time points: at baseline, after 3 and 6 months of intervention, respectively. Details of the study design and baseline characteristics of the participants have been published elsewhere( Reference Celis-Morales, Livingstone and Marsaux 3 ). The seven participating study centres were located in Munich (Germany), Athens (Greece), Dublin (Ireland), Maastricht (The Netherlands), Warsaw (Poland), Navarra (Spain) and Reading (UK), and recruitment was carried out countrywide. A total of 5562 participants (65 % females) were screened online over a 12-month period between August 2012 and August 2013 and consented to participate. Of these, 1607 (28·9 %) were recruited to the RCT. Participants aged 18–79 (mean 39·8 (sd 13·1)) years were included in the study, of whom 60·9 % (n 980) were women and 96·7 % were from white-European background. The mean BMI of all the participants was 25·5 (sd 5·2) kg/m2, and 44·8 % (n 721) of the participants were overweight or obese (BMI≥25·0 kg/m2). This study was conducted according to the guidelines laid down in the Declaration of Helsinki, and all the procedures involving human subjects were approved by the ethics committees of the participating centres. Written or online informed consent was obtained from all the participants. Further details on study design and participant characteristics can be found in the study by Celis-Morales et al.( Reference Celis-Morales, Livingstone and Marsaux 3 , Reference Livingstone, Celis-Morales and Navas-Carretero 19 ).
Samples and sampling protocol
Finger-prick blood samples were collected by participants themselves using a collection pack with two cards, one provided by Vitas Ltd, and one by DSM. Before spotting blood, cards for vitamin D analysis (Whatman Protein Saver 903 Card; GE Healthcare) were pre-treated with 1 % of 2,6-di-tert-butyl-4-methylphenol (BHT) dissolved in methanol (MeOH); 30 µl of 1 % BHT in MeOH were pipetted to each circle on the card and allowed to dry for at least 30 min at RT. These pre-treated cards were packed in an airtight Al bag (Whatman Foil Bags, item no. 10534321; Whatman Inc.) with a drying agent (Sorb-it, item no. 10548234; Süd-Chemie) and stored at RT until use. To help with blood collection, participants had access to an online video demonstration with written instructions and frequently asked questions in the language of recruitment. For the finger pricks, 2·0-mm contact-activated lancets (BD Microtainer; Becton, Dickinson and Company) were used. Each participant was asked to fill the two DBS cards each with five spots at each collection time point. This is the approximate equivalent of five drops of blood or 250 µl of blood/card. When the ten blood spots were filled, participants were instructed to let the cards dry at RT for at least 2 h, but not longer than 4 h, before samples were put back into the Al bags and returned by mail to the corresponding recruiting centre. The centres verified the content of the bags, and shipped one bag containing the DBS card to DSM (DSM Nutritional Products Ltd) for measurements of vitamin D (25(OH)D2 and 25(OH)D3). Although the shipments carried out done at ambient temperature, the closed bags were stored at the centres and at DSM at nominal −20°C.
Calibration was carried out using whole-blood samples received from blood donors of the ‘Blutspendezentrum SRK beider Basel’ (Blood Donation Centre at Basel Hospital), including haematocrit values for each sample. The calibration samples (n 15) had a mean haematocrit content of 44·2 % (range 38·7–49·3 %). Donors were twelve females and three males with an average age of 49 years. The calibration samples were prepared as follows: blood aliquots of 50 µl were pipetted onto a card and allowed to dry for 2–4 h at RT, avoiding exposure to direct sunlight; the cards were then transferred into Al foil bags with a desiccant inside and then stored in a freezer at nominal −20°C. Calibration samples were used for 3–6 months. As quality control, DBS samples with independently determined 25(OH)D3 content were analysed within each analytical run.
Reagents and instruments
25-Hydroxycholecalciferol monohydrate (25-OH-D3 monohydrate) was obtained from Dr Ehrenstorfer; 25-hydroxyergocalciferol (25(OH)D2) and SDS (≥99 %) were supplied by Sigma-Aldrich; 26,26,26,27,27,27-hexadeutero-25-hydroxycholecalciferol (25(OH)D3-d6) was supplied by Medical Isotopes; 2,6-di-tert-butyl-4-methylphenol (≥99 %, BHT), acetonitrile (gradient grade), formic acid (Suprapur), toluene and MeOH were from Merck; and MS-grade water was prepared using a Milli-Q instrument (Merck Millipore). Eppendorf tubes were centrifuged using a 5417 R model centrifuge (Vaudaux-Eppendorf), and evaporation was carried out using a Cyclone (Prolab). Analyte separation was performed using an Agilent 1260 HPLC with auto sampler, two binary pumps and column oven coupled with an AB Sciex Qtrap 5500 MS/MS-System with atmospheric pressure photoionisation (APPI source).
Assay
Sample preparation
Before analysis, the samples were assessed to check whether they met the quality criteria (Fig. 1): spot size (circle filled), thoroughly soaked (observed from the back) and one application of blood (not composed of many small spots). Two punches (inner diameter of 3·175 mm) were taken out of the card and placed into a 2-ml Eppendorf tube. After adding 100 µl of 0·1 % SDS solution and 20 µl of internal standard (ISTD, 25,25,25,26,26,26-hexa-deutero-25(OH)D3, 25 ng/ml in MeOH), the tubes were shaken at 40°C for 30 min. Subsequently, 400 µl of acetonitrile was added and the shaking continued at RT for 5 min (260/min, IKA shaker). Following this, the tubes were centrifuged at 20 000 g for 5 min, and the supernatant was transferred into a new 2-ml Eppendorf tube. The solvent was evaporated to dryness under vacuum (Cyclone), and the residue was reconstituted with 50-µl injection solvent (MeOH–water, 70:30) and transferred into a micro vial for analysis.

Fig. 1 Quality control criteria for dried blood spots from the Food4Me study. (a) Spot suitable for analysis. (b–d): Spots not suitable for analysis due to (b) small spot size not filling the circle, (c) multiple application of too small spots, including spots outside the circle, (d) multiple application of small spots and no thorough soaking of the paper (view from the back).
Chromatography and detection
Chromatography was performed using an Ascentis Express C18 column (Supelco), 7·5×2·1 mm, 2·7 µm, with a guard column using the following gradients: 0 min, flow 600 µl/min, 15 % A; 1·8 min, 600 µl/min, 0 % A; 1·9 min, 1000 µl/min, 0 % A; 3·7 min, 1000 µl/min, 0 % A; 3·8 min, 1000 µl/min, 15 % A; 5·8 min, 1000 µl/min, 15 % A; 5·9 min, 600 µl/min, 15 % A; and 6·0 min, 600 µl/min, 15 % A. Mobile phase A consisted of water containing 0·05 % formic acid; mobile phase B consisted of MeOH–acetonitrile (80:20, v/v) containing 0·05 % formic acid. Samples were kept at 10°C, the column temperature was 30°C and the injection volume was 10 µl. Dopant for APPI detection was toluene added post-column at 100 µl/min. Typical retention times were 1·55 min (25(OH)D3 and ISTD) and 1·71 min (25(OH)D2).
Detection was carried out using an AB Sciex 5500 Qtrap instrument with APPI positive mode and MRM scan type at unit resolution.
Parameter table: CUR 20; CAD Medium (9); IS 775; TEM 320; GS1 75; GS2 50; EP 10.
25(OH)D3, 383·3 Q1 Mass (Da), 211·3 Q3 Mass (Da), 50 Dwell (ms), 66 DP, 35 CE, 8 CXP; 25(OH)D3 qualifier, 383·3 Q1 Mass (Da), 257·2 Q3 Mass (Da), 50 Dwell (ms), 91 DP, 21 CE, 12 CXP; ISTD 25(OH)D3-d6, 389·3 Q1 Mass (Da), 211·3 Q3 Mass (Da), 50 Dwell (ms), 51 DP, 41 CE ,8 CXP; 25(OH)D2, 395·2 Q1 Mass (Da), 269·2 Q3 Mass (Da), 50 Dwell (ms), 46 DP, 29 CE, 12 CXP; 25(OH)D2 qualifier, 395·2 Q1 Mass (Da), 209·2 Q3 Mass (Da), 50 Dwell (ms), 56 DP, 39 CE, 10 CXP.
Determination of nominal 25-hydroxy vitamin D3 content of calibration samples
An aliquot of each whole-blood sample spotted for calibration was used for preparation of plasma. The 25(OH)D3 content of this plasma was measured using an established reference method( Reference Lauridsen, Halekoh and Larsen 20 ). The haematocrit content of the whole blood used for calibration was measured at the blood donation centre. As the haematocrit contents of the study samples were unknown, an estimated mean value of 40 % was used for calculation. This value was based on the rounded mean haematocrit content of the samples used for method development and validation. The following equation was used to normalise the calibration samples accordingly:
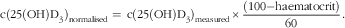 $${\rm c(25(OH)D}_{{\rm 3}} {\rm )}_{{{\rm normalised}}} = \, {\rm c(25(OH)D}_{{\rm 3}} {\rm )}_{{{\rm measured}}} {\times}{{(100{\minus}{\rm haematocrit})} \over {60}}.$$
$${\rm c(25(OH)D}_{{\rm 3}} {\rm )}_{{{\rm normalised}}} = \, {\rm c(25(OH)D}_{{\rm 3}} {\rm )}_{{{\rm measured}}} {\times}{{(100{\minus}{\rm haematocrit})} \over {60}}.$$
 $$$$
c(25(OH)D3)normalised: concentration of 25(OH)D3 (ng/ml plasma) in a calibration sample, normalised to 40 % haematocrit content; c(25(OH)D3)measured: concentration of 25(OH)D3 (ng/ml plasma) measured with reference method in plasma samples obtained from whole blood used for calibration; haematocrit: haematocrit value (%) for whole-blood sample obtained from Blutspendezentrum Basel.
$$$$
c(25(OH)D3)normalised: concentration of 25(OH)D3 (ng/ml plasma) in a calibration sample, normalised to 40 % haematocrit content; c(25(OH)D3)measured: concentration of 25(OH)D3 (ng/ml plasma) measured with reference method in plasma samples obtained from whole blood used for calibration; haematocrit: haematocrit value (%) for whole-blood sample obtained from Blutspendezentrum Basel.
For study samples, the resulting 25(OH)D3 concentration was then corrected for sex-specific mean haematocrit values of 41·5 % for female and 46·5 % for male participants by applying correction factors of 1·026 for females and 1·121 for males. This was based on information from the sex-specific reference ranges and means obtained from the seven clinical centres (U Hoeller et al., personal information).
Performance criteria
The method was validated based on the procedures described in the ‘Guideline on bioanalytical method validation’ of the European Medicines Agency( 21 ), taking into account specific requirements and recommendations for DBS analysis( Reference Timmerman, White and Globig 22 ). Selectivity was tested by comparing chromatograms from five samples of different donors with and without spike of either 25(OH)D3-d6 or 25(OH)D3 at 20 ng/ml blood. As the samples contained endogenous 25(OH)D3, area ratios of qualifier ion m:z (383:257) to quantifier ion m:z (383:211) were calculated and used for the assessment of selectivity of the detection of 25(OH)D3. Carry over was assessed by measuring blank samples after analysis of high-content samples. No significant carry over was observed. Linearity was determined by analysis of calibration samples according to the method on 3 different d. Linear regression of the analyte peak area ratio (analyte peak area v. ISTD peak area) against the nominal concentration was calculated. Differences between nominal concentrations and calculated concentrations were determined and expressed in percentage. Deviations of the measured values from nominal values should be ≤20 % and at least two-thirds of the calibration levels at each day should meet these criteria. As calibration was performed with endogenous samples, only a limited concentration range could be tested: 32·5–120 nmol/l calculated as plasma concentrations. The lower limit of quantification was set to 25 nmol/l, as this represents the accepted cut-off for vitamin D deficiency( Reference Bendik, Friedel and Roos 23 ). Accurate determination of lower concentrations was not within the scope of this study. For assessing accuracy, samples of five different subjects were analysed. Before spotting the whole-blood samples, plasma was separated from a whole blood aliquot of each sample and analysed using a reference method( Reference Bischoff-Ferrari, Dawson-Hughes and Stocklin 24 ). Nominal DBS concentrations were calculated by correcting the plasma concentrations with the corresponding haematocrit values (nominal concentration=plasma concentration×(100–haematocrit value)/100). DBS samples from each subject were analysed in 5-fold and compared with the nominal DBS concentration. Mean accuracy was calculated and reported as percentage of nominal DBS concentration value. As additional measure for accuracy and of the influence of varying haematocrit values in the calibration samples, we re-analysed calibration samples for which the 25(OH)D3 concentration values were assigned previously within a new set of calibration samples with independently assigned 25(OH)D3 concentration values. The mean haematocrit value of these samples was 42·0 (range 39·6–43·8) %. To determine intra-day precision, the results from samples prepared for accuracy were used. To estimate inter-day precision, triplicates of each sample were analysed on 3 different d. Precision was reported as CV of the measured concentrations. For determination of stability in the auto sampler at 10°C, extracts of incurred and spiked DSB samples were re-analysed after 60 h. Long-term stability was tested by storing DBS samples for upto 6 months at nominal −20°C and comparing the content analysed after the storage with initial values. There was good stability under both conditions.
Statistical analysis
Longitudinal linear mixed models were used to model 25(OH)D3 levels. Limit of detection (LOD) values in the 25(OH)D3 measurements were replaced by 12·5 nmol/l (four measurements, 0·1 %) and lower limit of quantitation (LOQ) values were replaced by 16·25 nmol/l 25(OH)D3 (134 measurements, 3·5 %). Sensitivity analyses were carried out assuming 0 nmol/l for LOD and LOQ values, and 25 nmol/l for LOD and LOQ values, respectively. For the centre with by far the most LOQ values (Dublin), the model fits differed by −4·0 nmol/l in winter, assuming 0 nmol/l for LOD and LOQ, and +0·2 nmol/l in summer. Assuming 25 nmol/l for LOQ and LOD values, the difference was +2·2 nmol/l in winter and −0·1 nmol/l in summer. The absolute deviations for all other centres were ≤2·4 nmol/l in winter and ≤0·2 nmol/l in summer.
To model the seasonal variation at the study sites, the study centre and the interaction of study centre with the functions sin(sample year×2×π) and cos(sample year×2×π) were included as fixed effects, and the participants as a random effect. The 20 January was found to be the consensus date across all study centres when the 25(OH)D3 concentrations reached their nadir. To simplify the subsequent modelling and interpretation, a single normalised sine function was derived, which oscillated between −1·0 when the 25(OH)D3 concentration was at its lowest on the 20 January and +1·0 when the 25(OH)D3 concentration was at its highest on the 21 July. It assumes synchronised timing of seasons across all study centres, but it differentiates mean levels and seasonal amplitudes by study centre. The function was coded as follows: standardised seasonal amplitude=SSA=sin((sample year−20/365·25−0·25)×2×π). On the 20th of each month, this function had the following values: −1·0 in January, −0·9 in February, −0·5 in March, 0·0 in April, 0·5 in May, 0·9 in June, 1·0 in July, 0·9 in August, 0·5 in September, 0·0 in October, −0·5 in November and −0·9 in December. For a sample taken on the 30 June, the sample year would equal 2013·5, and the resulting SSA would be 0·94. For each measurement, the corresponding SSA was thus calculated. The final model was then fitted using centre and the centre-SSA interaction as fixed effects and participant ID as random effect (presented in Table 2 and Fig. 2). Owing to the absence of a gold standard method, the biological and the analytical variability could not be separated in the present data set. The overall variation should thus be considered as a highly conservative estimate of the variation of the analytical method. R statistics software version 3.02 was used for all statistical analyses( 25 ), and within the R statistics software the function lme() in the package nlme( Reference Pinheiro, Bates and DebRoy 26 ) was used for the mixed model regressions.

Fig. 2 Individual measurements for 25-hydroxy vitamin D3 (25(OH)D3) by research centre and sampling date, as well as seasonal regression by centre. The model included 3711 measurements from 1412 participants. The predictors were the centre and the interaction of each centre with the standardised seasonal amplitude (SSA). The SSA for this data set is a sine function reaching its minimum −1·0 on 20 January and its maximum +1·0 on 21 July, as explained in the statistical methods section. The participant ID was included as random effect. The fixed effect regression fits are visualised within each plot. The largest seasonal oscillations were observed in Germany (92·1 nmol/l in summer v. 41·9 nmol/l in winter) and the smallest in Poland (67·1 nmol/l in summer v. 50·4 nmol/l in winter). Example calculation: on 20 May, the SSA reaches 0·5, therefore the estimate for a participant in Germany would be 67·0+0·5×25·1=79·6 nmol/l. Horizontal lines indicate vitamin D status intervals: <25 nm deficient, 25–50 nm insufficient, 50–75 nm sufficient, >75 nm optimal range.
Results
The assay described in the present study was developed for analysis of the large sample numbers from the Food4Me study. Calibration was performed using blood samples containing endogenous analyte content spotted on DBS cards. The nominal 25(OH)D3 concentration of these calibration samples was determined from plasma obtained from the same samples with an established reference method, corrected by the measured haematocrit values. Stability trials showed that these calibration samples could be used for at least 6 months if stored at nominal −20°C. The current method is equally applicable for determination of 25(OH)D2, but because substantial concentrations of 25(OH)D2 were not expected in the present study method validation focused on 25(OH)D3. An overview of the parameters validated and the results are given in Table 1. The re-analysis of calibration samples within an independent calibration showed a good correlation between the originally assigned 25(OH)D3 concentrations and the concentrations determined from the independent calibration for these samples (n 5; R 0·97). Overall, the DBS method showed results with slightly larger variations compared with current reference methodologies but within or close to the acceptance ranges of the guidelines. This led to specific characteristics of, for example, a rather small linear range of 32·5–120 nmol/l with deviation of upto 18 % between measured and nominal values (guideline limit 15 %) and precision upto 13–14 % CV for inter-day and intra-day. Overall, it was concluded that the method was suitable for determination of status levels for vitamin D, and thus for application in the Food4Me study.
Table 1 Overview of the performance characteristics of the analytical method

25(OH)D3-d6, 26,26,26,27,27,27-Hexadeutero-25-hydroxycholecalciferol; ISTD, internal standard.
In total, 3778 DBS samples were analysed: 453 from Germany, 560 from Greece, 554 from Ireland, 634 from The Netherlands, 530 from Poland, 555 from Spain and 492 from the UK. From 1003 participants, DBS at three time points could be measured, from 307 participants two time points could be measured, whereas for 155 participants only one time point could be measured. In addition, no samples were received for analysis from 259 participants (19·7 % of total cohort who completed the study); in addition, although a further sixty-seven samples gave valid results for 25(OH)D3 concentrations, they lacked a sampling date, and thus could not be included in the analysis of effects of seasons. Only eighty samples either did not meet the quality criteria or had insufficient blood spots for analysis of all the required parameters, and only five analyses failed due to technical errors. Overall, the quality of the samples was very good, showing that the methodology is suitable for unsupervised sampling at home following provision of detailed instructions to the participants including, for example, a video demonstration of the sampling process.
The data for 25(OH)D3 concentration are presented in Fig. 2. None of the samples contained 25(OH)D2 above the LOQ of 25 nmol/l. The seasonality of the 25(OH)D3 status for the participants of each study centre is clearly visible (P<0·001) with significantly higher concentrations in summer than in winter (67·1–92·1 v. 38·0–63·0 nmol/l). Details of the observed correlations are given in Table 2. Owing to the different starting dates, and the different duration of the study due to recruiting progress in each centre, the coverage of the annual time period varied between centres, but it usually included the seasonal minimum and maximum. DBS collection over more than 12 months occurred in only two centres – Dublin in Ireland and Reading in the UK. The values for the seasonal minima and maxima of 25(OH)D3 levels varied by country (Fig. 2). The largest seasonal changes were observed in the participants from Germany 92·1 nmol/l in summer v. 41·9 nmol/l in winter, whereas the smallest changes were recorded in Poland (67·1 v. 50·4 nmol/l, respectively). Overall, the highest values were found in late summer (21 July), and the lowest values were found in late winter (20 January). This is in good agreement with data from the literature, although the number of reports from comparable cohorts is limited( Reference Wahl, Cooper and Ebeling 27 ).
Table 2 Association of 25-hydroxy vitamin D3 (25(OH)D3) levels with the predictors ‘study centre’ and ‘seasonal amplitude’Footnote * (Coefficients with their standard errors)

* The study centre and the season per study centre were very good predictors of 25(OH)D3 levels. In Dublin, for example, the model estimated that the 25(OH)D3 levels oscillate by an amplitude of ±19·0 nmol/l around a mean of 57·1 nmol/l over the seasons. This translates to a minimum of 38·0 nmol/l on the 20 January, 57·1 nmol/l on the 20 April and a maximum of 76·1 nmol/l on the 21 July.
To assess the performance of the analytical method and the statistical model, a leave-one-out cross-validation was performed (Fig. 3). Only participants for whom all three planned measurements were available were included in the validation. For each measured value, a prediction was performed by taking into account the site and the season as fixed factors and the subject ID as a random factor. The two other measurements for the same participant were included each time, and the measurement to be predicted was excluded each time. In the absence of a gold standard, these predicted values were considered as the most plausible reference values against which the actual measurement values could be compared. The resulting Pearson’s correlation between measured and modelled values was r 0·65, and the differences between the modelled and measured values had an sd of 21·2 nmol/l. This variation is attributable both to biological and analytical variation.

Fig. 3 Comparison between modelled 25-hydroxy vitamin D3 (25(OH)D3) values and actually measured 25(OH)D3 values, based on a leave-one-out cross-validation of the model in Table 2 and Fig. 2. Only participants for whom all three pre-planned dried blood spots-based measurements were available were included in the validation. For each measured value, a prediction was performed by taking into account the site and the season as fixed factors and the subject ID as a random factor. Although the two other measurements of the same participant were included each time, the measurement to be predicted was excluded each time. The resulting Pearson’s correlation between measured and modelled values was r 0·65, and the differences between the modelled and measured values had an sd of 21·2 nmol/l.
Discussion
With 3778 valid measurements obtained from 1465 participants, to the best of our knowledge, the Food4Me study is the largest study using DBS samples where the participants did the sampling themselves at home. Sample preparation was designed to be as fast and as easy as possible, with the potential to perform all necessary steps in a multiwell plate. A crucial step for the whole methodology was the calibration of the assay. Although analysis of 25(OH)D3 from DBS has been applied before, we introduced improvements including calibration using DBS with endogenous analyte content and corrected by the haematocrit content of the calibration samples. As 25(OH)D3 is an endogenous compound, no blank samples of blood were available. In addition, spiking of blood with 25(OH)D3 was considered to be questionable, as the endogenous analyte is almost completely protein bound, and conditions to simulate this with spiking were difficult to achieve. Using blood with endogenous 25(OH)D3 content simplifies the calibration procedure and makes the methodology more robust as unknowns and calibration samples consist of the same matrix. As reference values for many micronutrients including vitamin D are typically reported as plasma levels, a conversion from whole blood is needed. This conversion is based on the haematocrit content that varies from person to person( Reference Thirup 28 ). As was the case in the Food4Me study, this value is often not available for study samples. Different approaches to circumvent this issue have been proposed( Reference De Kesel, Sadones and Capiau 29 ), which typically require an additional measurement of a second blood component such as K( Reference Capiau, Stove and Lambert 30 ), for normalisation, an approach that introduces additional analytical variance. For the present study, a haematocrit value of 41·5 % was assigned to samples from female subjects and a value of 46·5 % to samples from male subjects. As the study design did not include the concurrent sampling of venous blood and the analysis for 25(OH)D3 in the resulting plasma as a reference, the influence of haematocrit on the accuracy of the results could not be verified independently. However, we estimated that our assumption introduced a small amount of additional variation, perhaps <10 % – that is, when considering an exemplified normal range of 36–47 % haematocrit for women, a plasma concentration of 50 nmol/l calculated for a mean of 41·5 % haematocrit could vary between 45·7 and 55·2 nmol/l. The influence of other factors including spot size and location of punches on vitamin D determination in DBS have been reported in the literature( Reference Kvaskoff, Ko and Simila 31 ). Although our new method did not reach the accuracy of the reference analytics of 25(OH)D3 from plasma by LC-MS/MS, the results are comparable with, or better than, those reported for other DBS-based methods – for example, accuracy by recovery was reported in the range of 95·2–102·7 % (four concentration levels)( Reference Higashi, Suzuki and Hanai 12 ) and 80–118 % (three levels)( Reference Eyles, Anderson and Ko 9 ) and intra-assay precision in the range of 3·2–6·9( Reference Higashi, Suzuki and Hanai 12 ), 8–13( Reference Eyles, Anderson and Ko 9 ) and 11–13 % relative standard deviation (RSD) (three levels, inter-assay)( Reference Newman, Brandon and Groves 11 ). Overall, the new method was found to be suitable for status-level determination, as indicated by the performance criteria in comparison with international guidelines.
In the Food4Me study, sampling of the blood from the fingertip by participants was carried out unsupervised. This posed questions of compliance with the sampling protocol, and with regard to the impact of deviations from the protocol on overall sample quality. We found that the large majority of blood spots were of very good quality when assessed visually compared with our quality control criteria. This also suggests that this finger-prick approach to blood collection was well accepted by the participants, given the total of acceptable measurements, and that provision of both written and video instruction ensures that blood samples can be obtained reliably from untrained participants in large cohort studies across multiple countries. Quality aspects not assessable by visual inspection include too long or too short drying times of the blood and excessive sun or heat exposure of the sample before it reached the research centre. In addition, the identity of the blood donor could not be verified independently, although an embedded validation study of 10 % of participants was carried out, which included verification of identity based on genotype-provided reassurance on this question( Reference Celis-Morales, Livingstone and Woolhead 32 ). Therefore, the consistency of data points for each individual participant was used as an indirect quality indicator. The resultant sequential assessment of vitamin D status from three DBS measurements over 6 months is unique. A rapid increase in vitamin D status is possible in case of extended sun exposure or the use of high-dose supplementation, but a fast decline is very unlikely, because 25(OH)D3 has a half-life of approximately 20 d following oral administration( Reference Smith and Goodman 33 ). The demonstrated consistency of consecutive data from the individual participants proves the robustness of the methodology and the suitability of the DBS approach with unsupervised sampling for status determination of vitamin D3.
As expected, the results for all countries showed a strong seasonality with lowest mean vitamin D3 status levels observed towards the end of January and highest mean status levels observed towards the end of July. Although representative data for vitamin D status is available in some European countries( Reference Wahl, Cooper and Ebeling 27 ), data on seasonality are more limited. For the UK, lowest level of vitamin D3 concentrations were reported in February and the highest levels in September during the 2002–2004 period( Reference Hypponen and Power 34 ), with a difference of approximately 35 nmol/l. Furthermore, reports for Germany( Reference Zittermann, Scheld and Stehle 35 , Reference Hintzpeter, Mensink and Thierfelder 36 ) clearly demonstrate a seasonality with the highest median value in June and the lowest median value in March with a difference of 23·4 nmol/l for 1998( Reference Hintzpeter, Mensink and Thierfelder 36 ). Although the participants of the present study may not be representative of each country, our results are comparable with the published data. An in-depth analysis of differences between countries will be carried out separately.
Overall, the present study provided valuable insight into effects of seasonality of vitamin D3 status in 1465 participants from seven European countries. The DBS-based methodology of unsupervised sampling of DBS by the participants at home was found to be suitable for status determination of 25(OH)D3 in the setting of the large, international nutritional study Food4Me. This encourages application in future studies, and has the potential for simultaneous determination of other micronutrients from the same DBS samples.
Acknowledgements
This project was supported by the European Commission under the Food, Agriculture, Fisheries and Biotechnology Theme of the 7th Framework Programme for Research and Technological Development, grant number 265494.
The authors’ contributions are as follows: U. H. drafted the manuscript and developed the analytical method together with M. B. who carried out all the sample measurements; F. F. R. performed the statistical analysis; P. W. contributed to the design of the analytical methodology and data interpretation, J. C. M. was the study director of the proof-of-principle study of Food4Me; H. D., M. G., J. A. L., Y. M., J. A. M., W. H. M. S. and I. T. contributed to the design of the proof-of-principle study and were principle investigator for their respective research centre; L. B., R. F., H. F., E. R. G., M. G., K. H., S. K., C. P. L., K. M. L., A. L. M., C. F. M. M., C. C.-M., G. M., S. N.-C., C. B. O.’D., R. S.-C., A. S., L. T., M. C. W. and C. W. contributed to the study design and execution at the research centres. All authors contributed to, read and approved the final version of the manuscript.
U. H., M. B., F. F. R. and P. W. are employed by DSM Nutritional Products. The other authors have no potential financial or personal conflicts of interest to declare.





