


Colorectal cancer has become one of the leading causes of cancer-related deaths worldwide and is currently increasing in Western countries(Reference Jemal, Siegel and Ward1); consequently, much attention has been focused on preventive and therapeutic strategies. The beneficial effects of certain bioactive compounds, mainly present in fruits and vegetables, in preventing and treating colorectal cancer have become a major focus of research(Reference Cummings and Bingham2–Reference MacFarlane and Stover4). Among these, Bowman–Birk inhibitors (BBI) from legumes, such as soyabean (Glycine max), pea (Pisum sativum), lentil (Lens culinaris) and chickpea (Cicer arietinum), have been shown to have potential in protecting against inflammatory disorders and cancer development within the mammalian gastrointestinal tract(Reference Kennedy, Billings and Wan5–Reference Clemente, Sonnante and Domoney8). BBI are naturally occurring protease inhibitors with the ability to inhibit specifically serine (predominantly trypsin- and chymotrypsin-like) proteases. BBI are extensively disulphide-linked within proteins and have been demonstrated to be structurally and functionally resistant to the challenges (acidic conditions and the action of proteolytic enzymes) of the gastrointestinal tract in vivo. The conformational rigidity of BBI linked to the number and distribution of intramolecular disulphide bonds is mostly responsible for the high stability of these proteins towards extreme conditions and helps to maintain the structural and functional features of their binding loops(Reference Chen, Rose and Love9–Reference Trivedi, Laurence and Siahann11). BBI transit through the stomach and small intestine without major degradation, permitting significant amounts (5–8 % of total ingested) to reach the large intestine in active form(Reference Clemente, Jimenez and Marín-Manzano12). In addition, the biological activity of BBI is largely unaffected by the enzymatic and metabolic activity of faecal microbiota, retaining significance as a bioactive compound in the large intestine(Reference Marín-Manzano, Ruiz and Jimenez13).
In vitro and in vivo studies suggest that soyabean BBI can exert a protective and/or suppressive effect on cancer development within the gastrointestinal tract. Purified soyabean BBI and BBI concentrate, a protein extract of soyabean enriched in BBI(Reference Kennedy, Szuhaj and Newberne14), were both effective at concentrations as low as 10 mg/100 g of diet in reducing the incidence and frequency of colorectal tumours in the dimethylhydrazine rat model, without any adverse side effects documented for animal growth or organ physiology(Reference Kennedy, Billings and Wan5). Autoclaved BBI concentrate, in which the serine protease inhibitory activity of BBI was abolished, did not show any significant suppressive effect on colon tumour development, suggesting that the intrinsic ability of soyabean BBI to inhibit serine proteases may be required for their colorectal chemopreventive properties. Human epithelial cell lines derived from colon carcinoma are well-established models to investigate in vitro the action mechanism by which dietary compounds function in a chemoprotective role during the early stages of carcinogenesis. Recent studies have demonstrated a significant concentration- and time-dependent decrease in the growth of HT29 human colon adenocarcinoma cells in vitro, following treatment with soyabean BBI(Reference Clemente, Moreno and Marín-Manzano15). Similar effects have been reported for BBI from other legume sources, including those from pea(Reference Clemente, Gee and Johnson16) and lentil(Reference Caccialupi, Ceci and Siciliano17); the IC50 values (inhibitory concentration showing 50 % effect) for these protease inhibitors ranged from 32 to 73 μm. Interestingly, soyabean BBI that had been chemically inactivated did not demonstrate any significant effect on the proliferation of colon cancer cells, suggesting that a native conformation is necessary for BBI to exert anti-proliferative effects on such cells(Reference Clemente, Moreno and Marín-Manzano15).
In pea, the TI1 gene class encodes BBI that are expressed predominantly within seeds. TI1 is synthesised as a precursor protein of 114 amino acids, with a deduced mature peptide region of seventy-two amino acids having the ability to inhibit potently trypsin- and chymotrypsin-like proteases at the first and second inhibitory domains, respectively(Reference Domoney, Shewry and Casey18, Reference Domoney, Welham and Ellis19). In a previous study, we reported the inhibition of the growth in vitro of HT29 colon adenocarcinoma cells by a recombinant form of TI1 from the pea cv. Birte (rTI1B)(Reference Clemente, Gee and Johnson16). In the present study, we have evaluated the functional properties and anti-proliferative effects of rTI1B, expressed heterologously in Pichia pastoris, on colon cancer cells. We have compared the effects of rTI1B with those observed using a related mutant protein, obtained by site-directed mutagenesis of the P1 amino acid position in both inhibitory domains of TI1B. These novel data demonstrate the unambiguous relationship between the intrinsic ability of BBI to inhibit serine proteases and the associated negative effects on colon cancer cell growth, suggesting that serine proteases involved in early stages of carcinogenesis should be considered important targets in investigating the potential of BBI and related proteins as colorectal chemopreventive agents.
Experimental methods
Materials
Trypsin (type III) and α-chymotrypsin (type VII) from bovine pancreas, N-α-benzoyl-dl-arginine-p-nitroanilide (BAPNA), N-benzoyl-l-tyrosine ethyl ester (BTEE), high-glucose Dulbecco's modified Eagle's medium (DMEM), neutral red (NR) and additional high-grade chemicals for cell culture were obtained from Sigma. The P. pastoris host strain GS115 and the pPIC9 vector for expression and secretion of recombinant BBI were obtained from Invitrogen. The human colorectal adenocarcinoma HT29 and the normal colon fibroblastic CCD-18Co cell lines were supplied by the Cell Bank of the Scientific Instrumentation Centre at the University of Granada (CIC-UGR). Culture flasks and flat bottom ninety-six-well microtitre plates were purchased from Corning Costar and Nunc, respectively. All other chemicals were of analytical grade.
Growth media composition for Pichia pastoris
The P. pastoris GS115 (Invitrogen) host strain was maintained in yeast extract peptone dextrose broth (10 g yeast extract, 20 g peptone, 20 g dextrose per litre). Minimal dextrose plates (13·4 g yeast N base, 10 g glucose, 0·4 mg biotin and 15 g agar per litre) were used for the selection of putative transformants. Buffered minimal glycerol broth (13·4 g yeast N base, 0·4 mg biotin, 100 ml of 1 m-K2HPO4/KH2PO4 pH 6·0 and 1 % glycerol) was used until cultures reached log-phase growth. Buffered minimal methanol broth (identical to buffered minimal glycerol except for 0·5 % methanol instead of 1 % glycerol) was used to induce expression of the recombinant proteins.
Cloning, mutagenesis and expression of TI1B and derivative in the methylotrophic yeast Pichia pastoris
A sequence encoding a mature form of TI1B, a major BBI identified in pea seeds in vivo, was amplified from a cloned complementary DNA (cDNA) (pTI12-36)(Reference Domoney, Welham and Ellis19), using specific primers (N_peaTI1B and C_peaTI1B) (Table 1). The N and C primers incorporated XhoI and EcoRI cleavage sites, respectively, to allow directional cloning of the amplified DNA into the pPIC9 expression vector, used for in-frame integration of the TI1 gene into the P. pastoris genome. The N_peaTI1B primer was designed to encode a Kex2 processing site (Lys-Arg) followed by two Glu-Ala repeats, just before the N-terminus of TI1B, in order to increase the efficiency of Kex2 cleavage; an endogenous STE13 dipeptidyl aminopeptidase is predicted to remove both Glu-Ala dipeptides to yield the final recombinant proteins. PCR conditions were an initial denaturation step at 94°C for 5 min, followed by thirty-five cycles of 94°C for 30 s, 56°C for 30 s and 68°C for 1 min, and a final elongation step at 68°C for 5 min. The amplicon obtained was digested with both XhoI and EcoRI enzymes, ligated into pPIC9 vector previously digested with the same enzymes and used to transform Escherichia coli DH5α competent cells.
Table 1 List of primers used*

* The XhoI and EcoRI cleavage sites are underlined, the Kex2 processing site encoding Lys-Arg is in bold text, the Glu-Ala repeats are italicised and the mutations are in grey boxes. 5′AOX1 and 3′AOX1 are the sequencing primers.
In order to obtain an inactive TI1B-like protein, mutations were introduced by PCR into the active-site domains, using the overlap expression method(Reference Ho, Hunt and Horton20). The cDNA encoding TI1B was used as a template; the specific mutagenic primer pairs (F1-R1 and F2-R2, see Table1) were designed to introduce the amino acid Gly instead of Lys16 and Tyr42 at the P1 positions of the trypsin and chymotrypsin inhibitory domains, respectively. The N_peaTI1B and C_peaTI1B primers were used to amplify the final PCR amplified product, which was digested with XhoI and EcoRI, ligated into pPIC9 vector and used to transform DH5α cells as described previously. All PCR experiments were performed using a mixture of Platinum Pfx (Invitrogen) and MBL-Taq DNA polymerase (Dominion-MBL).
Plasmids containing inserts were selected with ampicillin (100 μg/ml medium) and the sequences verified by using the 5′AOX1 and 3′AOX1 primers (Table 1) on the pPIC9 vector. For efficient integration into the his4 locus of the P. pastoris genome, pPIC9-derived plasmids were first linearised by overnight digestion with SalI at 37°C, and digested DNA (5 μg) used to transform P. pastoris GS115 strain by electroporation (Gene Pulser XCell, Bio-Rad Laboratories). As a control, expression vector without any insert was used to transform P. pastoris. Transformants were selected on minimal dextrose agar plates without histidine, according to the Pichia expression kit manual (Invitrogen, a manual of methods for expression of recombinant proteins in P. pastoris, K1710-01). Up to sixteen individual colonies containing TI1B or its derivative mutant were grown overnight at 30°C in 100 ml of buffered minimal glycerol; cells were pelleted and resuspended in 20 ml of buffered minimal methanol. Incubation was carried out in an orbital shaker (250 rpm, 28°C) for 6 d, with methanol being added (0·5 %, v/v) every 24 h to compensate for consumption and evaporation. Strains secreting the highest levels of rTI1B or the corresponding mutant protein were selected for large-scale protein expression; selection was based on SDS-PAGE analysis and, where appropriate, trypsin inhibitor activity (TIA) and chymotrypsin inhibitor activity (CIA) assays (see the following text).
Protein expression and purification
The selected yeast transformants were inoculated in 40 ml of yeast extract peptone dextrose medium, cultured overnight at 28°C and then used to inoculate 4 litres of buffered minimal glycerol. When an OD600 nm in the range 2–6 was reached, cells were pelleted and resuspended in 800 ml of buffered minimal methanol medium. Cultures were then grown for 6 d at 28°C, and supplemented daily with 0·5 % (v/v) methanol to maintain the induced protein expression. Cells were pelleted and the supernatants (spent media) dialysed extensively against distilled water and freeze-dried. Protein extracts from freeze-dried dialysates were fractionated on a MonoS 5/50 GL cation exchange column (GE Healthcare), connected to an AKTA FPLC system (GE Healthcare), using a linear gradient of 0–0·22 m-NaCl in 50 mm-sodium acetate buffer, pH 4·4, at a flow rate of 1 ml/min. The elution was monitored at 280 nm and 0·5 ml fractions were collected. TIA measurements of eluted samples were carried out in flat-bottom microtitre plates and assay products measured at OD405 nm as previously described(Reference Clemente, Jimenez and Marín-Manzano12). CIA evaluation of eluted samples was carried out as described below. For rTI1B, column fractions containing inhibitory activity were pooled, dialysed extensively against distilled water and freeze-dried until use. In the case of the mutant form of rTI1B, where TIA and CIA were expected to be low or completely abolished, monitoring of the elution profile was carried out by SDS-PAGE.
Peptide mass fingerprinting and molecular mass determination of recombinant proteins
The recombinant proteins (10 μg) were dissolved in NuPAGE lithium dodecyl sulphate sample buffer (Invitrogen) and separated by electrophoresis on Novex 12 % Bis–Tris pre-cast gels using 2-N-morpholine-ethane sulphonic acid (NuPAGE MES, Invitrogen) as running buffer. Immediately before use, samples were reduced with dithiothreitol (DTT) and NuPAGE antioxidant added to the upper buffer chamber to prevent re-oxidation of reduced proteins during electrophoresis. Bands were excised from Colloidal Blue (Invitrogen)-stained gels and subjected to in-gel trypsin digestion. Peptide fragments from digested proteins were desalted and concentrated using C-18 ZipTip columns (Millipore) and then loaded directly onto the matrix-assisted laser desorption/ionization (MALDI) plate, using α-cyano-4-hydroxycinnamic acid as the matrix for MALDI-MS analysis. MS spectra were obtained automatically in a 4700 Proteomics Analyzer (Applied Biosystems) operating in reflectron mode with delayed extraction. Peptide mass data were used for protein identification against the MS protein sequence database (www.matrixscience.com). Additionally, a further attempt to identify the peptide mass data against the theoretical mass peptides derived from trypsin digestion of the rTI1B mutant, not included in databases, was carried out.
Freeze-dried purified proteins were dissolved in 1 % (v/v) trifluoroacetic acid (TFA), mixed with an equal volume of matrix (3 mg/ml of α-cyano-4-hydroxycinnamic acid in 70 % (v/v) acetonitrile/0·1 % TFA), deposited onto a MALDI sample probe and dried under ambient conditions. MS spectra were obtained automatically in a 4700 Proteomics Analyzer (Applied Biosystems), in the 800–10 000 m/z range, and average molecular masses of recombinant proteins were determined.
Measurement of protease inhibitory activities
rTI1B and its mutant derivative were assessed for TIA and CIA. TIA was measured using a modified small-scale quantitative assay with BAPNA as specific substrate, and using 50 mm-Tris, pH 7·5 as enzyme assay buffer. One trypsin inhibitor unit was defined as that which gives a reduction in absorbance at 410 nm of 0·01, relative to trypsin control reactions, in 10 min in a defined assay volume of 10 ml(Reference Domoney and Welham21). CIA was measured using BTEE as specific substrate. One chymotrypsin inhibitor unit was defined as that which gives a reduction in absorbance at 256 nm of 0·01, relative to chymotrypsin control reactions, in 5 min in a defined assay volume of 10 ml, as described previously(Reference Clemente, MacKenzie and Jeenes22). Specific TIA and CIA of the recombinant proteins, expressed as inhibitor units per mg of protein, were calculated. The inhibition constants (K i) of rTI1B for trypsin (pH 7·5) and chymotrypsin (pH 7·8), were determined from dose–response curves by competitive assays, using the chromogenic substrates BAPNA and BTEE, respectively(Reference Clemente, MacKenzie and Jeenes22). The reactions were initiated by adding trypsin (108 nm) or chymotrypsin (28 nm) with the respective substrate concentrations determined by K m measurements. The concentration of rTI1B required to achieve a half-maximal degree of inhibition (IC50) was determined for each protease, using the GraFit software (GraFit version 5, Erithacus Software Limited). K i were calculated from IC50 values using the tight-binding equations for competitive inhibitors(Reference Copeland, Lombardo and Giannaras23). The trypsin and chymotrypsin inhibitory patterns of rTI1B and its mutant derivative were analysed on 4-16 % zymogram blue casein gels (Invitrogen), in comparisons with the native proteins present in an albumin fraction from pea seeds(Reference Clemente, MacKenzie and Jeenes22). After electrophoresis, and following the manufacturer's instructions, gels were treated with zymogram renaturating buffer (Invitrogen) for 30 min at room temperature, equilibrated with zymogram developing buffer (Invitrogen), incubated with 25 ml of trypsin or chymotrypsin solution (0·08 mg/ml of zymogram developing buffer) at 37°C for 1·5 h, and washed with distilled water before the addition of acetic acid to stop the enzymatic reaction.
Cell viability assays
Human colorectal adenocarcinoma HT29 and normal colon fibroblastic CCD-18Co cells were maintained by serial passage in 75 cm2 plastic culture flasks. HT29 cells were cultured in DMEM, supplemented with fetal bovine serum (10 %), 2 mm-glutamine and 1 % antibiotic-antimicotic solution (Sigma, A5955), all at final concentration. In the case of CCD-18Co fibroblastic cells, media were supplemented additionally with non-essential amino acids to 1 % (final w/v; Sigma, M7145). Optimal assay conditions for rapidly dividing unpolarised, undifferentiated colonic cells were reported previously(Reference Clemente, Gee and Johnson16). The NR (3-amino-7-dimethylamino-2-methyl-phenazine hydrochloride) assay was validated by performing calibration assays, where HT29 and CCD18-Co cells were seeded in ninety-six-well microtitre plates at several cell densities. Accordingly, ninety-six-well microtitre plates were inoculated at a density of 2000 HT29 cells per well in 200 μl of growth media; in the case of CCD-18Co cells, 5000 cells per well were added. Plates were incubated under 5 % CO2 in humidified air for 24 h to allow the cells to adhere to the wells. rTI1B and its mutant derivative were dissolved in growth media at a range of concentrations (0–61 μm) and added to the cells under sterile conditions. Control cells received neither form of rTI1B. At the end of the growth period (96 h), the viability of HT29 and CCD-Co18 cells was assessed by the NR cytotoxicity assay, based on the ability of viable uninjured cells to incorporate and actively bind NR, a supravital dye, into lysosomes. Cells were stained with NR solution (2 h at 37 °C), followed by cell fixation (0·5 % formaldehyde, 0·1 % CaCl2 for 30 s) at room temperature. Plates were washed by two brief immersions in PBS (0·01 m-sodium phosphate buffer, 0·15 m-NaCl) and the dye extracted from the viable cells using an acidified ethanol solution (50 % ethanol, 1 % acetic acid overnight at 4°C). The absorbance of the solubilised dye was quantified at OD550 nm using a Bio-Rad Model 550 microplate reader (Bio-Rad). Cell viability data, expressed as a percentage of the values determined for control cells grown in the absence of either form of rTI1B, were obtained from at least three independent experiments (n ≥ 4 per experiment). The concentration of rTI1B that reduced cell viability by 50 % (IC50), as compared with untreated controls, was calculated by non-linear regression fit using the GraFit software. The data were analysed statistically by the Bonferroni's test to compare means and statistical significance was set at P < 0·05.
Results
Heterologous expression of TI1B and an engineered mutant
A cDNA clone (pTI12-36) was chosen for heterologous expression of TI1B, a major isoinhibitor from pea seeds, in P. pastoris. The in silico translation of the cDNA showed two inhibitory domains and fourteen cysteine residues in conserved positions, as described previously(Reference Domoney, Welham and Ellis19, Reference Domoney, Welham and Sidebottom24). Following the nomenclature of Schechter & Berger(Reference Schechter and Berger25), the residues Lys and Tyr at position P1 in the first (CTKSNPPTC) and second inhibitory domain (CAYSNPPKC), respectively, confer specificity for inhibition of trypsin and chymotrypsin-like proteases. To obtain an inactive TI1B-like protein, pTI12-36 cDNA was used as a template and mutations to its active sites achieved by site-directed mutagenesis. Overlapping PCR was used to substitute the P1 amino acid within the trypsin (Lys16) and chymotrypsin (Tyr42) inhibitory domains with a Gly at both positions.
Pichia pastoris was selected for the expression of rTI1B and its mutant derivative since this host had been shown previously to be capable of folding and secreting a BBI from lentil(Reference Caccialupi, Ceci and Siciliano17). Sequences corresponding to the pea cDNA, pTI12-36, encoding either a mature form of TI1B lacking any of the forty-two amino acid prepropeptide sequence, or the mutant derivative of TI1B predicted to be inactive, were cloned into the vector pPIC9 downstream of the α-factor secretion signal sequence to direct the recombinant proteins into the secretory pathway of P. pastoris. Expression of both recombinant forms of TI1B was induced by methanol, and the recombinant proteins were subsequently secreted into the culture supernatants. TIA and CIA assays were used for rapid screening of a large number of putative TI1B transformants after induction with methanol; spent media from transformants expressing the mutant form of rTI1B lacked inhibitory activity in these assays (not shown). In the culture supernatants of putative transformants of rTI1B and its mutant derivative, polypeptides in the range 7–9 kDa were detected by SDS-PAGE; in the case of yeast transformed with the empty vector, these polypeptides were absent from culture supernatants (not shown). Yeast transformants expressing the highest levels of rTI1B were selected by TIA and CIA for subsequent large-scale protein expression, whereas transformants secreting the rTI1B mutant were selected by SDS-PAGE followed by protein concentration determination, as previously described(Reference Clemente, MacKenzie and Jeenes22). Zymography of culture supernatants of rTI1B under non-denaturing conditions allowed the detection of two bands with specific inhibitory activities against the digestive enzymes trypsin and chymotrypsin (Fig. 1). These bands showed an increased mobility on non-denaturing gels when compared with the primary mature product of the TI1 gene observed in vivo. In zymograms, the overall charges of the rTI1B forms were observed to be − 3 and − 4, whereas that deduced from the sequence of the mature TI1B variant in vivo is − 2(Reference Domoney, Shewry and Casey18). Culture supernatants from the rTI1B mutant derivative failed to inhibit in-gel activity of either digestive enzyme (Fig. 1).

Fig. 1 In-gel protease inhibitory activity analyses of recombinant TI1B (rTI1B) and its inactive mutant. Zymogram blue casein gels were treated with the digestive enzymes, trypsin (T) or chymotrypsin (C); dark areas indicate where the enzyme has been inhibited. Lane 1: pea seed albumin fraction from cv. Birte; lane 2: culture supernatant from secreted expression of rTI1B; lane 3: culture supernatant from secreted expression of the mutant rTI1B predicted to be inactive. Lane 1 contained 1·5 mg of pea albumin fraction, whereas lanes 2 and 3 contained 20 μg of recombinant protein. The direction of electrophoresis on non-denaturing gels is indicated (vertical arrow), alongside the overall charge of the major isoinhibitors found in pea seeds of cv. Birte. Among the isoforms present in seeds, the − 2 variant represents the primary mature product of the TI1B gene (horizontal arrow).
Purification, molecular and functional characterisation of the recombinant proteins
To purify the recombinant proteins, secreted proteins were fractionated by MonoS cation exchange chromatography. The elution patterns of rTI1B and its inactive mutant are shown in Fig. 2(A) and (B), respectively. The recombinant proteins were retained by the Mono S column at pH 4·4. In good agreement with results obtained from non-denaturing gels, rTI1B was resolved as two chromatographic peaks, which eluted in the range 0·07–0·10 m-NaCl (peak 1) and 0·13–0·16 m-NaCl (peak 2), and the two chromatographic peaks showed both TIA and CIA. The rTI1B mutant derivative (see later for positive identification) was eluted as a single broad chromatographic peak in the range 0·04–0·09 m-NaCl, having neither TIA nor CIA (Fig. 2(B)). Differences in the elution pattern of the two recombinant proteins can be predicted to be due to the substitution of Lys16 at the P1 position of the trypsin inhibitory domain with Gly in the rTI1B mutant. The chromatographic fractions containing the recombinant proteins were pooled and analysed by SDS-PAGE; in all cases, purified fractions showed proteins in the range 7–9 kDa. The peak 2 from rTI1B showed a reduced gel migration when compared to peak 1, with the latter possibly representing a processed variant (Fig. 2(C)). Further studies by MALDI-time of flight (TOF) MS and mass peptide fingerprinting were carried out in order to determine the authenticity of the recombinant proteins. The experimentally determined m/z values of peaks 1 and 2 derived from rTI1B were 7694 and 8146 Da, respectively, clearly differing from the value predicted for mature TI1B, allowing for disulphide formation (7946 Da) (Fig. 3). In the case of the inactive mutant, a mixture of three differentially processed isoinhibitors was deduced, based on molecular masses of 7386, 7515 and 7968 Da, compared with an expected mass of 7768 Da for the mutant construct with correct disulphide bond formation. The presence of these three forms was not revealed by SDS-PAGE; this may be due to differences in their relative proportions. Given their lack of inhibitory activity, further chromatographic separation of inactive forms was not carried out before cell assays. In addition, the amino acid sequences of the two recombinant proteins were matched by mass peptide fingerprinting (Fig. 3). In-gel tryptic digestion of excised electrophoretic bands was performed followed by separation of the peptides generated and MS-based analysis. A search of peptide mass data against the MS protein sequence database enabled the unambiguous identification of TI1B (Swiss-Prot entry: IBBA_PEA). The sequence coverage of peaks 1 and 2 from rTI1B was 86 and 75 %, respectively. An attempt to identify the peptide mass data against the theoretical mass peptides derived from trypsin digestion of the rTI1B mutant, not included in databases, was also carried out. Sequence coverage of the inactive mutant was 82 % and included both inhibitory domains having their corresponding Gly substitution. Although endoproteolytic removal of the signal sequence by the P. pastoris Kex2 protease clearly occurred, differences in mobility on non-denaturing gels (Fig. 1), molecular mass determination and mass peptide fingerprinting data (Fig. 3) supported the existence of a Glu-Ala extension (200 Da) at the N-terminus of the recombinant proteins (Fig. 3); this indicates an inefficiency in cleavage by the endogenous STE13 dipeptidyl aminopeptidase(Reference Damaso, Almeida and Kurtenbach26, Reference Chen, Wang and Cong27). Recombinant forms having the complete Glu-Ala-Glu-Ala extension or those fully processed by STE13 were not observed. In the case of peak 1 from rTI1B, a loss of four amino acids (Val Ile Lys Asn) at the C-terminus was deduced whereas, in the case of the inactive mutant, a loss of an additional amino acid (Glu Val Ile Lys Asn) was deduced for one isoform, compared with those predicted from the DNA sequences (Fig. 3). Both an N-terminal extension (Glu Ala) and deduced processing at the C-terminus, involving loss of charged amino acid(s), are in good agreement with the observed mobility on non-denaturing gels of rTI1B (peaks 1 and 2) when compared with the primary mature product of the TI1 gene observed in vivo (Fig. 1). The mass and peptide data obtained for the mutant protein are consistent with correct protein folding, based on disulphide bond formation, and the m/z variants are consistent with differential processing of C-terminal sequences, as indicated in Fig. 3. All further experiments, including kinetic and cells assays, were performed with unprocessed rTI1B (peak 2) and the mixture of isoinhibitors derived from the inactive mutant (Fig. 3).

Fig. 2 Elution profile of (A) recombinant TI1B (rTI1B) and (B) its mutant derivative on a MonoS 5/50 GL cation exchange column. Absorbance (mAU) at 280 nm of the chromatographic elution and the linear gradient of NaCl (0–0·22 m) are shown (solid and dotted lines, respectively). Using N-α-benzoyl-dl-arginine-p-nitroanilide and N-benzoyl-l-tyrosine ethyl ester as specific substrates, the trypsin (▲) and chymotrypsin (Δ) inhibitory activities, measured on every fraction are shown. Where there was little or no inhibition (B), then monitoring of the elution profile was carried out by SDS-PAGE (gel inset in B); (C) SDS-PAGE under denaturing and reducing conditions of the inactive mutant protein (lane 2) and peaks 1 and 2 from rTI1B (lanes 3 and 4, respectively), following the chromatography steps. Molecular weight (MW) markers are shown in lane 1.

Fig. 3 Molecular mass determination and amino acid sequences deduced by peptide mass fingerprinting of recombinant TI1B (rTI1B) and its mutant derivative, compared with the primary mature pea protein encoded by the TI1B gene (Q41065, Swiss-Prot database, Swiss Institute of Bioinformatics). Amino acid sequences of inhibitory domains are underlined. P1-P1′ are the reactive peptide bond sites, in bold text. Lys (K) and Tyr (Y) at position P1 determine specificity for trypsin and chymotrypsin, respectively. Amino acid sequences of recombinant proteins were deduced from mass peptide fingerprinting analyses; the peptide sequences determined for recombinant proteins are italicised. The molecular masses of the recombinant variants, determined by MALDI-TOF, are shown compared with their predicted values. The sequence Glu Ala Glu Ala was engineered just before the N-terminus of recombinant proteins for efficient cleavage by Pichia Kex2. The deduced sequences of the recombinant proteins indicate an inefficient cleavage of the dipeptide Glu Ala by the endogenous STE13 dipeptidyl aminopeptidase.
The ability of rTI1B to inhibit the digestive enzymes trypsin and chymotrypsin was evaluated (Table 2). The specific TIA and CIA of this recombinant protein were 2476 (sd 238) and 2956 (sd 196) units per mg of protein, respectively. The rTI1B mutant was unable to inhibit either enzyme. Based on IC50 and K i calculations, rTI1B was demonstrated to be a potent inhibitor of trypsin (K i of 21 (sd 2) nm) and chymotrypsin (K i of 8 (sd 1) nm) (Table 2). Such values fall within the nanomolar range reported previously for various members of the BBI family, including those from soyabean(Reference Clemente, Moreno and Marín-Manzano15), lentil(Reference Caccialupi, Ceci and Siciliano17) and lupin (Lupinus albus)(Reference Scarafoni, Consonni and Galbusera28), and suggested that TI1B was expressed as a correctly folded active recombinant protein from P. pastoris.
Table 2 Inhibition constant (K i) and specific inhibitory activity (IU) for trypsin (T) and chymotrysin (C) of recombinant TI1B (rTI1B) and its mutant derivative*
(Mean values and standard deviations from at least five independent determinations)
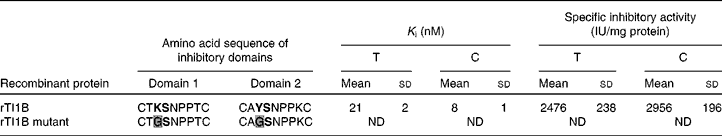
ND, not detected.
* The reactive peptide bond sites are marked in bold text. K determines specificity for trypsin, whereas Y determines specificity against chymotrypsin. Amino acid substitutions in the rTI1B mutant are highlighted in grey.
Effect of rTI1B and the corresponding inactive mutant on the proliferation of human colon cells
The effects of the recombinant proteins on the growth of human colon adenocarcinoma HT29 cells were determined by comparing the cell viability of cultured cells in the absence or presence of rTI1B or the corresponding mutant protein (0–61 μm), monitored by the cytotoxic NR cell assay. At concentrations greater than 15 μm, rTI1B inhibited the in vitro growth of HT29 cells in a concentration-dependent manner (Fig. 4(A)); the IC50 value for rTI1B was 31 (sd 7) μm, in agreement with those obtained for BBI from plant sources, including lentil(Reference Caccialupi, Ceci and Siciliano17) and soyabean(Reference Clemente, Moreno and Marín-Manzano15). In contrast, the growth of HT29 cells was not reduced when treated with the rTI1B related mutant, even at the highest concentration tested (61 μm). In addition, the growth of non-malignant CCD-18Co colon cells was unaffected by either rTI1B or its inactive variant in the range of concentrations tested (0–61 μm, Fig. 4(B)).
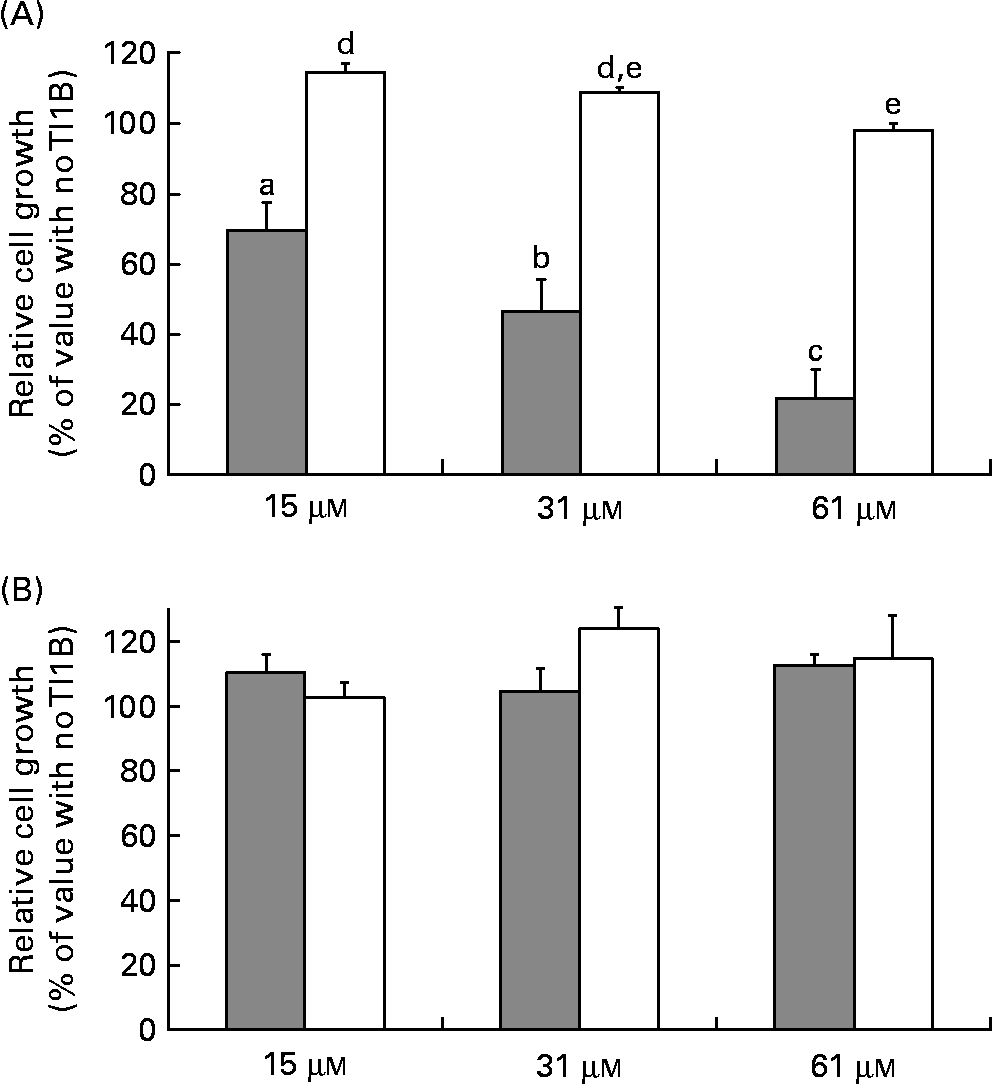
Fig. 4 Effects of recombinant TI1B (rTI1B, closed bars) and the corresponding mutant protein (open bars) on the in vitro growth of (A) HT29 human colorectal adenocarcinoma cells and (B) normal colon fibroblastic CCD-18Co cells. Growth media were supplemented with protein in the concentration range 0–61 μm and cells harvested after a period of 96 h. Values are means, with standard deviations represented by vertical bars of at least three independent experiments, each having four technical replicates. a,b,c,d,e Mean values with different letters were significantly different (P <0·05; Bonferroni's test).
Discussion
Among other dietary constituents, soyabean BBI and homologous proteins appear to be promising chemopreventive agents within the gastrointestinal tract(Reference Kennedy, Billings and Wan5). These dietary constituents, present at high concentrations in legume seeds, have been shown to be effective at preventing or suppressing radiation- and chemical carcinogen-induced transformation, in a wide variety of in vitro assays, and carcinogenesis in in vivo model systems, as reviewed elsewhere(Reference Clemente and Domoney6–Reference Clemente, Sonnante and Domoney8, Reference Kennedy29, Reference Kennedy30). In previous studies, we have demonstrated a significant concentration- and time-dependent decrease in the growth of an array of colon cancer cells (HT29, LoVo, Caco2) when treated with BBI proteins from different sources, including soyabean(Reference Clemente, Moreno and Marín-Manzano15), lentil(Reference Caccialupi, Ceci and Siciliano17) and pea(Reference Clemente, Gee and Johnson16); after soyabean BBI treatment, HT29 colon cancer cells showed a dose-dependent increase in the proportion of cells that were arrested in the G0-G1 phase(Reference Clemente, Moreno and Marín-Manzano15). It was shown furthermore that treatment of soyabean BBI with reducing and alkylating agents, which substantially reduces inhibitory activity against serine proteases, renders these dietary proteins unable to inhibit cell proliferation of colon cancer cells(Reference Clemente, Moreno and Marín-Manzano15). Treatments like autoclaving(Reference Kennedy, Billings and Wan5) or chemical modification via alkylation of sulphydryl groups(Reference Clemente, Moreno and Marín-Manzano15) have been used as tools for probing the association between protease inhibition and anti-cancer properties of BBI. Related studies have indicated the importance of disulphide bonds in maintaining the three-dimensional structure and inhibitory activity of BBI(Reference Singh and Appu Rao31). Where severe disruptive treatments are used, the native conformation of BBI is lost, alongside losses of tryptic and chymotryptic inhibitory activities, making the relationship between protease inhibition, protein structure and health-beneficial effects unclear. In the present study, we have provided new evidence to support a relationship between the anti-proliferative properties of BBI and their specific inhibition of serine proteases. Here, a loss of native conformation and/or correct protein folding is avoided by comparing the anti-proliferative cellular properties of a major pea isoinhibitor, TI1B, capable of inhibiting both trypsin- and chymotrypsin-like proteases, with a related mutant predicted to lack both TIA and CIA. Both proteins were expressed in recombinant form by heterologous expression in the methylotrophic P. pastoris system, capable of efficiently folding extensively disulphide-bonded proteins, as reported for lentil BBI(Reference Caccialupi, Ceci and Siciliano17) and other unrelated protease inhibitors(Reference Volpicella, Ceci and Cordewener32). In this study, the masses of the secreted recombinant proteins were consistent with correct disulphide bond formation. When treated with rTI1B, a significant concentration-dependent decrease in the growth of HT29 human colon adenocarcinoma cells was observed, whereas non-malignant colonic fibroblast CCD-18Co cells were unaffected; in contrast, the rTI1B mutant lacking TIA and CIA did not exert any significant effect on the growth of HT29 cells (Fig. 4). These findings suggest that serine proteases should be considered as molecular targets in investigating the potential chemopreventive role of BBI and related proteins during the early stages of carcinogenesis.
Yavelow et al. (Reference Yavelow, Collins and Birk33) reported that an enzymatically modified BBI having only CIA was still fully effective as an inhibitor of radiation-induced transformation in vitro, whereas the modified inhibitor with TIA only was ineffective. Although the modified BBI may have been impaired in the inhibition of several molecular targets compared with the native protein, these early observations led to the hypothesis that chymotrypsin-like proteases are potential targets of BBI in anti-cancer effects. In contrast, it has been demonstrated recently that a major soyabean BBI isoinhibitor which inhibits trypsin-like proteases only exerts anti-proliferative properties against colon cancer cells(Reference Clemente, Moreno and Marín-Manzano15). These data suggest that both trypsin- and chymotrypsin-like proteases involved in the early stages of carcinogenesis should be considered as potential targets of BBI-like proteins. So far, the therapeutic targets and the action mechanisms of BBI remain unknown. However, the present study shows that the successful expression of individual isoinhibitors, both naturally occurring and engineered BBI, can assist in understanding the mechanism by which these dietary proteins can inhibit cancer cell proliferation, and in identifying and validating their precise therapeutic targets(Reference Clemente and Domoney6, Reference Clemente, Sonnante and Domoney8). In parallel, direct comparative serine protease profiling studies of malignant and non-malignant cells will explain the differential effects on cell proliferation observed for the variant rTI1B tested here, contributing to the assessment of BBI for preventive and/or therapeutic medicine.
Acknowledgements
A. C. acknowledges support by ERDF-co-financed grants from the Spanish CICYT (AGL 2007-60007 and AGL2010-15877) and from CSIC (PIE-200970I054). C. D. acknowledges support from the European Union (Grain Legumes Integrated Project, a Framework Programme 6 project, grant no. FOOD-CT-2004-506223) and from Defra (grants nos AR0105 and AR0711). The authors are very grateful to Dr S. Ogueta and A. Lario from the Proteomics Facility of University of Cordoba (Spain) and the Institute of Parasitology and Biomedicine Lopez Neyra (Granada, Spain), respectively, for carrying out peptide mass fingerprinting and molecular mass determination. This study is not subject to any conflicts of interest. A. C. and C. D. were responsible for the experimental design, data analysis, interpretation and writing of the manuscript. M. C. M.-M. was responsible for gene cloning, site-directed mutagenesis and heterologous expression of recombinant proteins. E. J. and M. C. A. carried out protein purification and enzymatic kinetic experiments.






