

Microdeletion syndromes have been found to be associated with various psychiatric comorbidities, such as hyperkinetic disorder, autism spectrum disorders, obsessive–compulsive disorders for individuals diagnosed with Prader–Willi syndrome (PWS) (Reference Dykens, Hodapp and WalshDykens 1992), or schizophrenia and schizoaffective disorders for individuals with 22q11.2 deletion syndrome (22q11DS) (Reference Gothelf, Frisch and MichaelovskyGothelf 2009). The prevalence of intellectual disabilities has been estimated to be approximately 1–3% in high- and middle-income countries. This may result in marked psychosocial impairments, particularly for those with moderate, severe and profound intellectual disabilities. Microdeletion is considered to be one of the prenatal causes of intellectual disability. Other prenatal causes include central nervous system infections, exposure to radiation, maternal use of various substances and drugs, as well as malnutrition.
Microdeletions are alterations of the chromosomes that cannot be detected by conventional light microscopy or other conventional methodologies, and are usually approximately 1–3 Mb long. Although the site and size of the deletion may vary for a particular syndrome, it should be noted that a particular area of a critical gene is usually involved. Given that critical areas of gene are affected, the deletion often results in characteristic phenotypic changes due to the lack of expression of particular segments of genes. To date, the visualisation of these deletions has only been possible via methods such as comparative genomic hybridisation or fluorescence in situ hybridisation (FISH). Such consistent alterations in DNA content have been associated with some well-established malformation syndromes (Reference SchinzelSchinzel 1988). In circumstances in which psychiatrists suspect the presence of an underlying genetic disorder, it is thus recommended for the individual to undergo genetic testing. Visualisation of the deletions would be done using the methodologies described above.
The most common microdeletion syndromes are listed in Box 1, together with the other known microdeletion syndromes. Table 1 gives an overview of the common syndromes. The latest microdeletion syndrome to be discovered is the 8p23.1 syndrome, which involves the duplication of a region of chromosome 8. The phenotypic changes include changes in speech, developmental delays as well as cardiovascular-related disorders.
BOX 1 Microdeletion syndromes
Most common microdeletion syndromes
-
15q11-13 maternal deletion syndrome (Angelman syndrome)
-
15q11-13 paternal deletion syndrome (Prader–Willi syndrome)
-
7q11.23 deletion syndrome (Williams syndrome)
-
4p deletion syndrome (Wolf–Hirschhorn syndrome)
-
Cri du chat syndrome
-
DiGeorge syndrome
-
Rubinstein–Taybi syndrome
Other microdeletion syndromes
-
1q36 deletion syndrome
-
1q21.1 deletion syndrome
-
Thrombocytopenia-absent radius syndrome
-
2q15-16.1 deletion syndrome
-
2q23.1 deletion syndrome
-
2q37 deletion syndrome
-
3p deletion syndrome
-
3q29 deletion syndrome
-
5q35 deletion syndrome (Sotos syndrome)
-
6p25 deletion syndrome
-
8p23.1 deletion syndrome
-
8q22.1 deletion syndrome (Nablus mask-like facial syndrome)
-
8q24.11 deletion syndrome (Langer– Giedion syndrome or trichorhinophalangeal syndrome type II)
-
9q22 deletion syndrome
-
9q34.3 deletion syndrome
-
DiGeorge syndrome/velocardiofacial syndrome complex 2
-
11q13 deletion syndrome (WAGR syndrome)
-
11q11.2 deletion syndrome (Potocki–Shaffer syndrome)
-
11q24.1 (Jacobsen syndrome)
-
13q14 deletion syndrome (retinoblastoma syndrome)
-
15q11.2 deletion syndrome (BP1-2)
-
15q13.3 deletion syndrome
-
15q15.3 deletion syndrome
-
15q24 deletion syndrome
-
16p13.11 deletion syndrome
-
17p13.3 deletion syndrome
-
17p1.2 deletion syndrome
-
17q12 deletion syndrome
-
17q21.31 deletion syndrome
-
18p deletion syndrome
-
20p11 deletion syndrome
TABLE 1 Summary of the core features of each of the common microdeletion syndromes
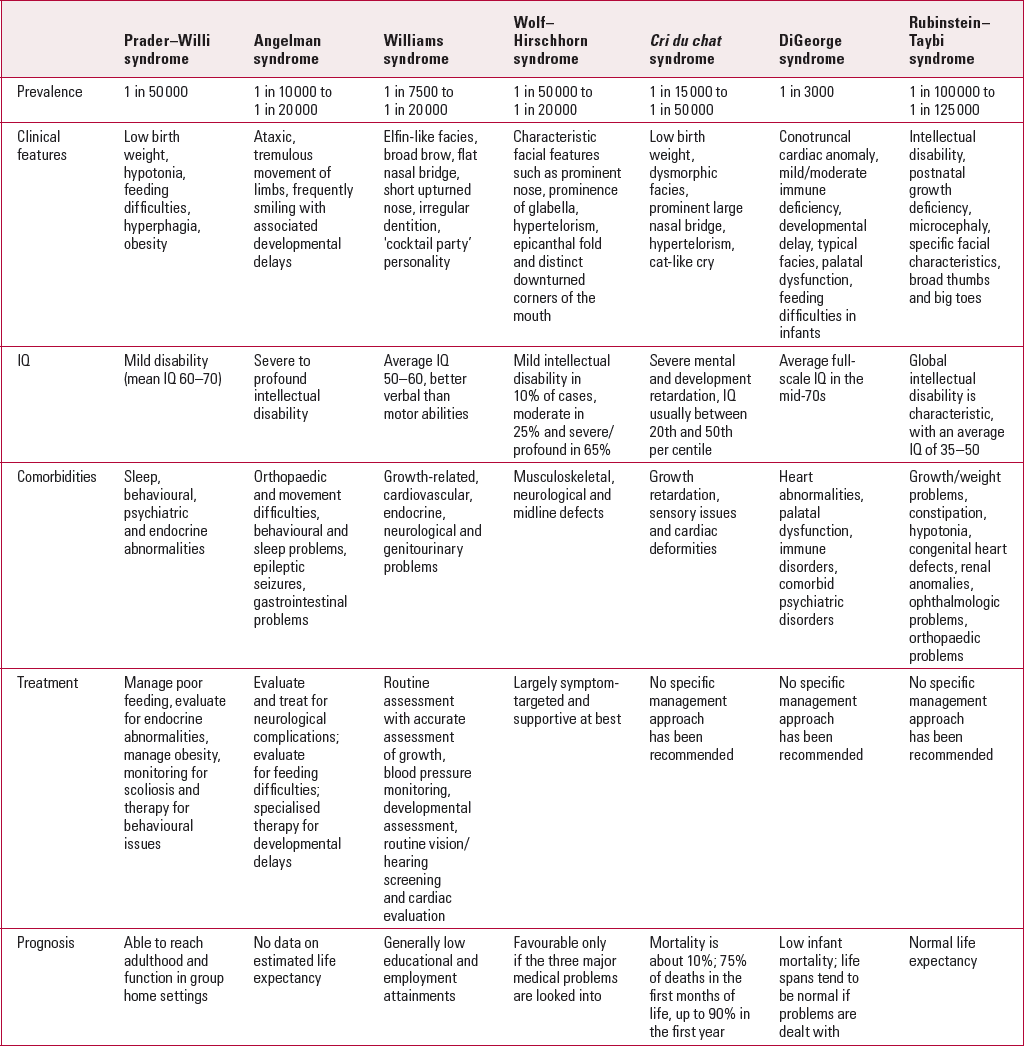
It is hoped that this review article will provide the psychiatrist with an update about the clinical features of each of the common microdeletion syndromes and their association with intellectual disabilities, and share recent insights and updates regarding clinical management and genetic testing for each of the individual syndromes.
Chromosome 22q11.2 deletion syndrome
Background
This syndrome (22q11DS) refers to patients with a hemizygous deletion of chromosome 22q11.2. The syndrome is well recognised worldwide and commonly known as either DiGeorge syndrome or velocardiofacial syndrome. The hemizygous deletion is inherited in an autosomal dominant pattern. Hence, affected parents have a substantial risk of passing on the deletion to their children; 22q11DS is estimated to occur in nearly 1 in 3000 children.
Clinical features
The clinical features of 22q11DS are diverse, but common features include a conotruncal cardiac anomaly, mild-to-moderate immune deficiency, developmental delays, palatal dysfunction, and feeding difficulties in infants (Reference ShprintzenShprintzen 2008). Of note, patients with 22q11DS do not tend to have noticeable facial dysmorphisms (Reference ShprintzenShprintzen 2008). More importantly, 22q11DS is commonly associated with congenital heart disease; the deletion is found in a large percentage of children with conotruncal heart anomalies (Reference Goldmuntz, Clark and MitchellGoldmuntz 1998). Palatal dysfunction is also relatively common, with 8% of children with cleft palates, submucous cleft palate and occult submucous cleft palates having 22q11DS (Reference Shprintzen, Siegel-Sadewitz and AmatoShprintzen 1985). Despite the fact that immune disorders are also relatively common, few affected individuals have severe immunodeficiency (Reference ShprintzenShprintzen 2008).
Psychiatric comorbidity and intellectual disabilities
Speech and language impairment is commonly seen, with approximately 75% of affected individuals having hypernasal speech and a high percentage having severe articulation impairment (Reference ShprintzenShprintzen 2008). Speech development is usually mildly delayed and receptive language abilities exceed expressive abilities (Reference Golding-Kushner, Weller and ShprintzenGolding-Kushner 1985).
Psychiatric disorders seen in children and adolescents with 22q11DS include attention-deficit hyperactivity disorder (ADHD; 35–46%) (Reference Niarchou, Martin and ThaparNiarchou 2015), oppositional defiant disorder (16–43%), specific and social phobias (23–61%), generalised anxiety disorder (GAD; 17–29%), separation anxiety disorder (16–21%), obsessive– compulsive disorder (OCD; 4–33%), major depressive disorder and dysthymia (10–20%) and autism spectrum disorders (14–45%) (Reference Tang, Antshel and FremontTang 2015). By late adolescence and adulthood, up to one-third of patients with 22q11DS develop psychotic disorders resembling schizophrenia and schizoaffective disorders (Reference Gothelf, Frisch and MichaelovskyGothelf 2009). Individuals diagnosed with schizophrenia in the general population tend to have a premorbid period in which they have gradual worsening of their social and academic functioning. Individuals with 22q11DS tend to have a more chronic and prolonged duration of impaired functioning, from childhood to adolescence (Reference Radoeva, Fremont and AntshelRadoeva 2017).
The average full-scale IQ score in affected individuals is in the mid-70s, within the borderline intelligence range (Reference Swillen, Devriendt and LegiusSwillen 1997); 25 to 40% score in the intellectual disability range. The cognitive profile of individuals with this condition is characterised by relative strengths in the areas of reading, spelling, and rote memory, and relative weaknesses in the areas of visuospatial memory and arithmetic (Reference Simon, Bish and BeardenSimon 2005).
Prognosis
With improved palliative cardiac repair and medical management of immunodeficiency, infant mortality in 22q11DS is approximately 8% (Reference Ryan, Goodship and WilsonRyan 1997). Individuals with 22q11DS can have normal lifespans if they undergo corrective cardiac procedures. The immune problems for some of these individuals also tend to remit over time. The associated endocrinology problems can be treated with appropriate medications (Reference ShprintzenShprintzen 2008).
Prader–Willi syndrome
Background
PWS is a complex multisystem genetic disorder that is caused by the lack of expression of paternally inherited imprinted genes on chromosome 15q11-q13 (Reference JinJin 2011). The main gene implicated is SNRPN (small nuclear ribonucleoprotein N). This encodes a protein involved in pre-mRNA splicing and processing in brain and muscle (Reference YoungYoung 2005). Approximately 70% of cases result from 4-Mb microdeletions involving the paternally derived chromosome 15. Approximately 25% of cases result from maternal uniparental disomy (UPD) for chromosome 15. The remaining cases are due to a mutation or tiny deletion in the PWS imprinting centre or SNRPN promotor. The prevalence of PWS varies from 1 in 8000 to 1 in 20 000, with population prevalence at approximately 1 in 50 000 (Reference JinJin 2011).
Clinical features
The common clinical features of PWS include low birth weight, severe hypotonia and feeding difficulties in early infancy, followed by hyperphagia and obesity starting in early childhood (Reference BuitingBuiting 2010). Common comorbidities include sleep abnormalities, behavioural and psychiatric disturbances, and endocrinological abnormalities. Individuals with PWS are predisposed to sleep-disordered breathing, including central and obstructive sleep apnoea, abnormal arousal, abnormal circadian rhythms in rapid eye movement (REM) sleep, reduced REM latency, and abnormal response to hypercapnia, as well as excessive daytime sleepiness (Reference Nixon and BrouilletteNixon 2002). PWS has been found to be associated with endocrinological abnormalities such as obesity, growth hormone deficiency and hypogonadism.
Psychiatric comorbidity and intellectual disabilities
Previous research has found that children with PWS tend to have behavioural and psychiatric disturbances, which usually include behavioural patterns such as temper tantrums, obsessive– compulsive characteristics, autism spectrum disorder, and attention-deficit hyperactivity symptoms (Reference Dykens, Hodapp and WalshDykens 1992). Previous research has found a close association between this microdeletion syndrome and bipolar disorder (Reference Boer, Holland and WhittingtonBoer 2002; Reference Verhoeven, Tuinier and CurfsVerhoeven 2003). Based on a prior population-based study of 25 individuals aged 18 or more, it was ascertained that 28% have an underlying bipolar disorder and the medium age of onset is approximately 26 years (Reference Boer, Holland and WhittingtonBoer 2002). A combination of medications such as antipsychotics and mood stabilisers have been used in the treatment of those diagnosed with bipolar disorder (Reference Boer, Holland and WhittingtonBoer 2002).
In addition, previous research has highlighted that most individuals with PWS are diagnosed with mild intellectual disability, with a mean IQ of 60–70. However, 20% of individuals with PWS are diagnosed as having moderate intellectual disability (Reference JinJin 2011).
Clinical management
Care for patients with PWS includes management of hypotonia or poor feeding, evaluation for endocrine abnormalities, management of obesity, monitoring for scoliosis and therapy for behavioural issues. With regards to the management of the hyperphagia and binge eating behaviours, pharmacological approaches such as use of anorectic agents have not shown any effectiveness (Reference Martin, State and KoenigMartin 1998). Psychiatric medications such as selective serotonin reuptake inhibitors might regulate the behavioural symptoms commonly seen in patients with PWS, but these do not mediate the binge eating and weight gain symptoms (Reference Martin, State and KoenigMartin 1998). Psychotropic drugs could be initiated to help with behavioural management (Reference Martin, State and KoenigMartin 1998).
Prognosis
Patients with PWS frequently reach adulthood and are able to function in a group home setting, performing vocational work or attending community college classes. Complications from hypogonadism (e.g. osteoporosis/pathological fracture), behavioural issues (e.g. temper tantrums and stubbornness) and morbid obesity (e.g. type 2 diabetes mellitus and cor pulmonale) may shorten life expectancy and affect the quality of life (Reference JinJin 2011).
Angelman syndrome
Background
Angelman syndrome involves microdeletion of the maternally derived chromosome 15 in approximately 70% of cases, and paternal UPD for chromosome 15 in 2–3% of cases. Most commonly, the microdeletion involves the 15q12 site. Most of the remainder of cases are caused by mutations in UBE3A (ubiquitin-protein ligase E3A) or another gene involved in the imprinting process. Severe psychomotor retardation is attributed to abnormal expression of UBE3A, which encodes a protein ligase thought to play an important part in the localisation of proteins in the brain (Reference YoungYoung 2005). The incidence of Angelman syndrome is estimated to be between 1 in 10 000 and 1 in 20 000 (Reference WilliamsWilliams 2005).
Clinical features
Patients with Angelman syndrome tend to have ataxia and tremulous limb movements. They tend to have a happy demeanour with sudden bursts of laughter. In addition, they often have developmental delays (Reference Williams, Beaudet and Clayton-SmithWilliams 2006). Some of the common physical features of the syndrome include hypotonia, microbrachycephaly, vertical inclination of skull base, occipital furrow, fair hair, ocular abnormalities, midface hypoplasia, macrostomia and widely spaced teeth, tongue protrusion with drooling, and prognathism (Reference SchinzelSchinzel 1988). The most common comorbidities include orthopaedic and movement difficulties, behavioural issues and sleep problems, epileptic seizures and gastrointestinal problems (Reference Larson, Shinnick and ShaayaLarson 2015).
Psychiatric comorbidity and intellectual disabilities
Individuals with Angelman syndrome tend to have severe to profound intellectual disabilities, with developmental delays that are first noted at around 6 months of age. They tend to be able to sit unsupported at only 12 months of age, crawl at the age of 18–24 months and walk at the age of 4 years (Reference Clayton-SmithClayton-Smith 1993). They tend to have limited vocabulary of only one or two words, despite having reasonable comprehension of simple commands and sentences (Reference Laan, van Haeringen and BrouwerLaan 1999). It should be noted that the results of any neuropsychological tests might be confounded by the patient's inability to focus, underlying hyperactivity and inability to communicate (Reference Williams, Driscoll and DagliWilliams 2010).
Prior research has highlighted that another common comorbid condition in Angelman syndrome (in addition to intellectual disability) is autism spectrum disorder (Reference Ornoy, Weinstein-Fudim and ErgazOrnoy 2016). Individuals with class I deletions, which involve multiple breakpoints in chromosome 15q11-13, were found to have a higher incidence of autism spectrum disorder.
Clinical management
Eighty per cent of patients with Angelman syndrome have neurological complications such as epilepsy. Early screening is therefore essential, as are further evaluations such as electroencephalogram (EEG) to check for the presence of seizures. In addition, in view of the developmental delays as well as the communication difficulties, patients should be referred for speech therapy. For patients with gait and movement problems, physical and occupational therapy should be considered. With regards to sleep disturbances, medications such as melatonin could be considered.
Prognosis
There are no data on the life expectancy for patients with Angelman syndrome. It is to be expected that patients with Angelman syndrome would continue to have epileptic seizures throughout adulthood, with atypical absence seizures and myoclonic seizures being the most prominent. With advancing age, adults tend to become less active, largely due to joint contractures, thus leading to significant difficulties with walking. Some individuals eventually require a wheelchair (Reference Buggenhout and FrynsBuggenhout 2009).
Williams syndrome
Background
Williams syndrome is caused by a ~1.5-Mb deletion encompassing the elastin gene (ELN) at 7q11.23 (Reference Morris and MervisMorris 2000). The deletion results in the loss of approximately 28 genes, including ELN itself. The estimated prevalence of Williams syndrome ranges from 1 in 7500 to 1 in 20 000 (Reference Martens, Wilson and ReutensMartens 2008).
Clinical features
Individuals with Williams syndrome have a characteristic elfin facies, with a broad brow, flat nasal bridge, short upturned nose, wide mouth with full lips, and irregular dentition (Reference Morris, Mervis, Goldstein and ReynoldsMorris 1999). They often have an engaging ‘cocktail party’ personality which conceals mild intellectual disability (Reference YoungYoung 2005). The most common comorbidities include growth-related problems, and cardiovascular, endocrine, neurological and genitourinary abnormalities.
Psychiatric comorbidity and intellectual disabilities
Individuals with Williams syndrome are noted to have mild-to-moderate developmental delay. In neuropsychological testing, it is consistently noted that they tend to have higher verbal intelligence scores compared with their performance intelligence scores. The average IQ for patients with Williams syndrome lies between 50 and 60 (Reference Martens, Wilson and ReutensMartens 2008). Hence, it is not uncommon to find that these individuals have better verbal abilities as compared to motor abilities (Reference Martens, Wilson and ReutensMartens 2008). Recent studies have found increased prevalence of autism spectrum disorders among patients with Williams syndrome (Reference Ornoy, Weinstein-Fudim and ErgazOrnoy 2016). Clinicians need to be cognisant of the fact that the microdeletion in Williams syndrome could lead to a range of social communications deficits, ranging from lack of verbal language to being excessively talkative (Reference Tordjman, Anderson and BotbolTordjman 2012).
Clinical management
It is recommended that a multidisciplinary team follows up the patient, owing to the numerous complications associated with Williams syndrome. The American Academy of Pediatrics guidance (Committee on Genetics 2001) previously recommended evaluations based on the patient's age. In general, the recommendations emphasise the importance of routine physical assessment with accurate assessment of growth, as well as blood pressure monitoring, developmental assessment, routine vision and hearing screening, and – of paramount importance – routine cardiac evaluation.
Prognosis
Owing to the multitude of complications (both physical and psychiatric) associated with Williams syndrome, individuals with this condition have generally low educational and employment attainments (Reference Howlin and UdwinHowlin 2006).
Wolff–Hirschorn syndrome
Background
Patients with Wolff–Hirschorn syndrome (WHS) have a deletion of variable size involving the short arm of chromosome 4, always including the band 4p16. Eighty per cent of deletions arise de novo, while 20% occur as a result of a parentally transmitted unbalanced translocation (Reference YoungYoung 2005). The gene affected is the WHS candidate 1 gene, which has a role in the transcription process (Reference Baradaran-Heravi, Lange and AsakuraBaradaran-Heravi 2013). The incidence is estimated at 1 in 50 000 to 1 in 20 000 births, with a female predilection of 2:1 (Reference Battaglia, Filippi and CareyBattaglia 2008).
Clinical features
Individuals with WHS can be easily identified, as they tend to have a characteristic facial appearance, with a prominent nose due to a high forehead, prominence of the glabella, hypertelorism, epicanthal fold, and a distinct mouth with downturned corners (Reference Battaglia, Filippi and CareyBattaglia 2008). This characteristic facial appearance is commonly known as the ‘Greek warrior helmet’ appearance. Other common clinical features include musculoskeletal and neurological abnormalities, as well as midline defects. The most commonly associated medical problems are seizures, feeding difficulties, and cardiac, ophthalmological and dental abnormalities (Reference Battaglia, Filippi and CareyBattaglia 2008).
Psychiatric comorbidity and intellectual disabilities
Global developmental delay is often exhibited, with mild intellectual disability in 10%, moderate in 25% and severe/profound in 65% of cases. Expressive language, although limited to guttural or disyllabic sounds in most individuals, was at the level of simple sentences in 6%. Comprehension seems to be limited to a specific context. Individuals’ abilities to communicate gradually improve over time as they learn more gestures to facilitate communication (Reference Battaglia, Filippi and CareyBattaglia 2008).
Clinical management
Clinical management is largely symptom targeted and supportive at best. It is essential to note that patients diagnosed with WHS are prone to recurrent aspirations, and hence they are prone to recurrent episodes of respiratory infections. In view of the expected difficulties with feeding, there have been recommendations for the consideration of placement of a gastrostomy tube. In view of the presence of midline defects, routine thorough cardiovascular examination is required. In addition, in view of the presence of ptosis and hearing defects, it might also be wise to conduct audiological and ophthalmological screening periodically.
Prognosis
The prognosis of patients with WHS is considered to be favourable if the three major medical problems (seizures, feeding difficulties and developmental disabilities) are carefully investigated and managed (Reference Battaglia, Filippi and CareyBattaglia 2008). If diagnosed early, cardiac, ophthalmological and dental abnormalities can be treated with good outcomes (Reference Battaglia, Filippi and CareyBattaglia 2008).
Cri du chat syndrome
Background
Cri du chat syndrome (CdCS) is a genetic disorder resulting from a deletion of variable size occurring on the short arm of chromosome 5 (5p−). This rare disease has an incidence ranging from 1 in 15 000 to 1 in 50 000 live-born infants (Reference MainardiMainardi 2006). CdCS affects less than 1% of the population with mild intellectual disability; its prevalence has been previously reported as 1 in 350 among 6000 individuals with intellectual disability (Reference NiebuhrNiebuhr 1978).
Clinical features
Patients with CdCS are noted to have low weight at birth (mean 2614 g). In addition, they may have dysmorphic facies, characterised by microcephaly, round face, prominent large nasal bridge, hypertelorism, epicanthal folds, downward slanting palpebral fissures, downturned corners of the mouth, low-set ears, micrognathia, abnormal dermatoglyphics (transverse flexion creases) and the typical cat-like cry (Reference JonesJones 2006). The phenotypic features of the cat-like cry have been localised to the region of 5p15.3 (Reference Mainardi, Perfumo and CalìMainardi 2001). It is important for clinicians to note that these features might become less prominent with advancing age, which could cloud the clinical diagnosis; in particular, it appears that the face tends to become longer and narrower (Reference MainardiMainardi 2006). In addition, patients with CdCS may have growth retardation, sensory issues and cardiac deformities.
Psychiatric comorbidity and intellectual disabilities
Patients with this condition tend to have severe mental and developmental retardation. The IQ is usually below the 20th and 50th per centile, respectively. In a study of 26 UK children with CdCS, full-scale IQ on the Wechsler Intelligence Scale for Children (WISC-III) varied from below 40 (4 children) to between 40 and 57 (the remaining children), with a mean of 48 (Reference Cornish, Bramble and MunirCornish 1999). Neuropsychological testing has revealed that patients tend to have better abilities in comprehension of speech compared with their ability to communicate (Reference MainardiMainardi 2006).
Prognosis
After the first year of life, survival rates are high and morbidity is low. It has been found that mortality is about 10%, with 75% of premature deaths occurring in the first months of life, and up to 90% within the first year (Reference NiebuhrNiehbur 1978).
Rubinstein–Taybi syndrome
Background
Rubinstein–Taybi syndrome (RSTS) generally occurs sporadically, and 55% of affected individuals show a microdeletion of chromosome 16p13.3, or a mutation in the gene encoding either CREB-binding protein or EIA-binding protein. However, these mutations cannot be found in 45% of individuals with the syndrome, leaving the diagnosis to rest on clinical features alone (Reference HennekamHennekam 2006). The syndrome is rare, with the birth prevalence ranging from 1 in 100 000 to 1 in 125 000 (Reference HennekamHennekam 2006).
Clinical features
RSTS is well defined by several congenital anomalies, including intellectual disability, postnatal growth deficiency, microcephaly, specific facial characteristics, broad thumbs and big toes (Reference Rubinstein and TaybiRubinstein 1963). Patients with RSTS have a striking facial appearance, with highly arched eyebrows, long eyelashes, down-slanting palpebral fissures, broad nasal bridge, beaked nose with the nasal septum extending well below the alae, highly arched palate, and mild micrognathia. Their facial expression tends to have a grimace or unusual smile with almost closing of the eyes. They also often have talon cusps at the permanent incisors, and broad thumbs and broad big toes (Reference HennekamHennekam 2006). Common comorbidities include growth/weight problems, constipation, hypotonia, congenital heart defects, renal anomalies, problems with anaesthesia, ophthalmologic problems, orthopaedic problems, increased risk for tumours, seizures, behavioural issues and intellectual disability (Reference HennekamHennekam 2006).
Psychiatric comorbidity and intellectual disabilities
Affected individuals have several behavioural issues, including short attention span and poor coordination, as well as sudden mood changes in adulthood (Reference HennekamHennekam 2006). In addition, recent studies have highlighted the association between this disorder and characteristic behaviours such as motor stereotypies (Reference Galera, Taupiac and FraisseGalera 2009). Other studies have demonstrated an age-related change in symptoms, with anxiety, mood instability and aggression being more prominent as age advances (Reference Yagihasi, Kosaki and OkamotoYagihasi 2012). However, individuals are usually described as loving, friendly and happy as children (Reference Stevens, Hennekam and BlackburnStevens 1990). Global intellectual disability is characteristic, with an average IQ between 35 and 50 (Reference Hennekam, Baselier and BeyaertHennekam 1992).
Prognosis
Survival rates in general are good, with apparently normal life expectancy and frequent reports of adults with RSTS (Reference HennekamHennekam 2006).
Prevention of microdeletion syndromes and associated disabilities
Prevention of microdeletion syndromes can be done via DNA-based prenatal diagnosis. The purpose of DNA-based prenatal diagnosis is usually to determine whether the fetus has inherited the disease-causing mutation(s) identified in one or both parents. It is also led by ultrasound findings that may occasionally suggest a specific disorder, such as investigations for decreased fetal motor activity in PWS (Reference SchinzelSchinzel 1986).
Karyotyping remains the gold standard of chromosome analysis; however, this technique has limited resolution and is therefore unable to reliably detect small discrete chromosomal abnormalities involving genomic regions of less than 6–10 Mb, such as microdeletions. With the advances in cytogenetics, the development of the FISH technique makes this possible. Fetal cells can be extracted from amniotic fluid samples and analysed using FISH. However, the majority of FISH assays are highly targeted and will only uncover abnormalities for which specific probes have been designed.
As such, these DNA-based prenatal diagnosis methods are mostly targeted towards families at risk of an inherited condition, and are not used as a screening test because of cost, technical complexity and limited resources. The family should first undergo molecular genetic testing and genetic counselling before embarking upon a DNA-based prenatal diagnosis (Reference BuiBui 2002; Reference Konialis, Hagnefelt and SevastidouKonialis 2011).
With regards to the secondary disabilities that might arise as a complication of the underlying microdeletion syndrome, other rehabilitation options may be useful, such as physiotherapy and speech and language therapies.
Conclusions
This article has highlighted the association between common microdeletion syndromes and intellectual disabilities. Genetic testing would help in early detection, so that appropriate clinical management plans could be formulated to help individuals better manage the other complications arising from microdeletion syndromes.
MCQs
Select the single best option for each question stem
-
1 The estimated incidence of intellectual disability owing to microdeletion syndromes in high- and middle-income countries is:
-
a 1–3%
-
b 4–5%
-
c 5–7%
-
d 7–10%
-
e More than 10%.
-
-
2 Schizophrenia occurs most frequently in:
-
a Prader–Willi syndrome
-
b Angelman syndrome
-
c Williams syndrome
-
d cri du chat syndrome
-
e 22q11.2 deletion syndrome.
-
-
3 IQ of 50–60, with higher verbal than performance intelligence score, is commonly found in:
-
a Prader–Willi syndrome
-
b Angelman syndrome
-
c Williams syndrome
-
d cri du chat syndrome
-
e 22q11.2 deletion syndrome.
-
-
4 The second most common cause of developmental delay and the major cause of congenital heart disease after Down syndrome is:
-
a Prader–Willi syndrome
-
b Angelman syndrome
-
c Williams syndrome
-
d cri du chat syndrome
-
e 22q11.2 deletion syndrome.
-
-
5 Common comorbidities for individuals with 22q11.2 deletion syndrome do not include:
-
a hyperkinetic disorder
-
b oppositional defiant disorder
-
c enuresis
-
d separation anxiety disorder
-
e obsessive–compulsive disorder.
-
MCQ answers
| 1 | a | 2 | e | 3 | c | 4 | e | 5 | c |


eLetters
No eLetters have been published for this article.