



LEARNING OBJECTIVES
After reading this article you will be able to:
• identify the range of genetic tests available and how these apply to intellectual disability, autism spectrum disorder and epilepsy
• decide when to refer to a clinical geneticist
• know where to find information resources on genetic testing.
Neurodevelopmental disorder encompasses a heterogeneous group of conditions including intellectual disability, autism spectrum disorder (ASD), attention-deficit hyperactivity disorder (ADHD), speech and language disorders, specific learning disorders and motor disorders.
Underlying differences in brain development and function that contribute to neurodevelopmental disorders can be caused by a wide range of environmental and genetic factors. The factors that influence brain development and function are not specific to neurodevelopmental disorders and may also result in other medical conditions, such as epilepsy and sensory impairments affecting vision and hearing. Genetic variation that leads to neurodevelopmental differences can also contribute to a wide range of multisystem conditions. Enquiries into coexisting physical conditions are therefore important as they can be essential for interpretation of genetic findings.
This article focuses on advances in genomic medicine in the UK and the current understanding of the role of genomics in neurodevelopmental disorders.
Background
The field of genomics has moved swiftly in the past couple of decades, with the National Human Genome Project completing the first whole sequence of the 3.2 billion base pairs of the human genome in April 2003. Following on from this, UK research completed through the Deciphering Developmental Disorders study (Box 1) and the 100 000 Genomes Project (Box 2) has highlighted the high level of variation and complexity of genetics within rare disorders (including neurodevelopmental disorders) and cancer.
BOX 1 The Deciphering Developmental Disorders (DDD) project
Recruitment commenced in April 2011 and was completed in April 2015.
The DDD project continues to discover new genes for developmental disorders and provide diagnoses.
The study brought together doctors in the 24 regional genetics services throughout the UK and Republic of Ireland, with scientists at the Wellcome Sanger Institute, a charitably funded research institute which played a world-leading role in sequencing (reading) the human genome:
• 13 500 UK families were recruited to the study
• over 4500 diagnoses were made
• over 180 research papers have been published.
Supported by patient support groups:
• SWAN UK (syndromes without a name)
• Unique (rare chromosome disorders).
Information available at www.ddduk.org
BOX 2 The 100 000 Genomes Project
Recruitment commenced in 2013 and was completed in December 2018
The 100 000 Genomes Project was set up by Genomics England, a company wholly owned and funded by the Department of Health and Social Care, with the aims of bringing benefit to patients and setting up a genomic medicine service within the NHS:
• 100 000 genomes sequenced
• 85 000 NHS patients affected by rare disease or cancer
• 18.5% of data turned into actionable findings
• diagnostic yields for intellectual disability, hearing impairment and vision disorders ranged from 40 to 55%
• increase in diagnostic yield across a range of rare diseases which in 25% of cases has led to support in management and clinical decisions (The 100 000 Genomes Project Pilot Investigators 2021).
Information available at www.genomicsengland.co.uk/initiatives/100000-genomes-project
The innovative infrastructure established during the development of the 100 000 Genomes Project has pioneered the future of genomic services. NHS England is working in partnership with Genomics England to create a robust system to embed genomics into mainstream clinical care to provide personalised medicine.
To oversee and implement this, the National Health Service (NHS) launched its Genomic Medicine Service (GMS) in England in 2018 to establish common standards and protocols, and a uniform approach to testing has been achieved in England with the introduction of the National Genomic Test Directory. Alongside this, Health Education England's Genomics Education Programme is supporting the development and upskilling of the healthcare workforce.
Preparing for change
A literature review published in 2019 looked at improving access to genetic testing for adults with intellectual disability (Adlington 2019). The paper highlighted the need for education to update professionals on advances in genetic techniques and raised ethical issues regarding communication and consent in individuals with reduced mental capacity (Fig. 1).
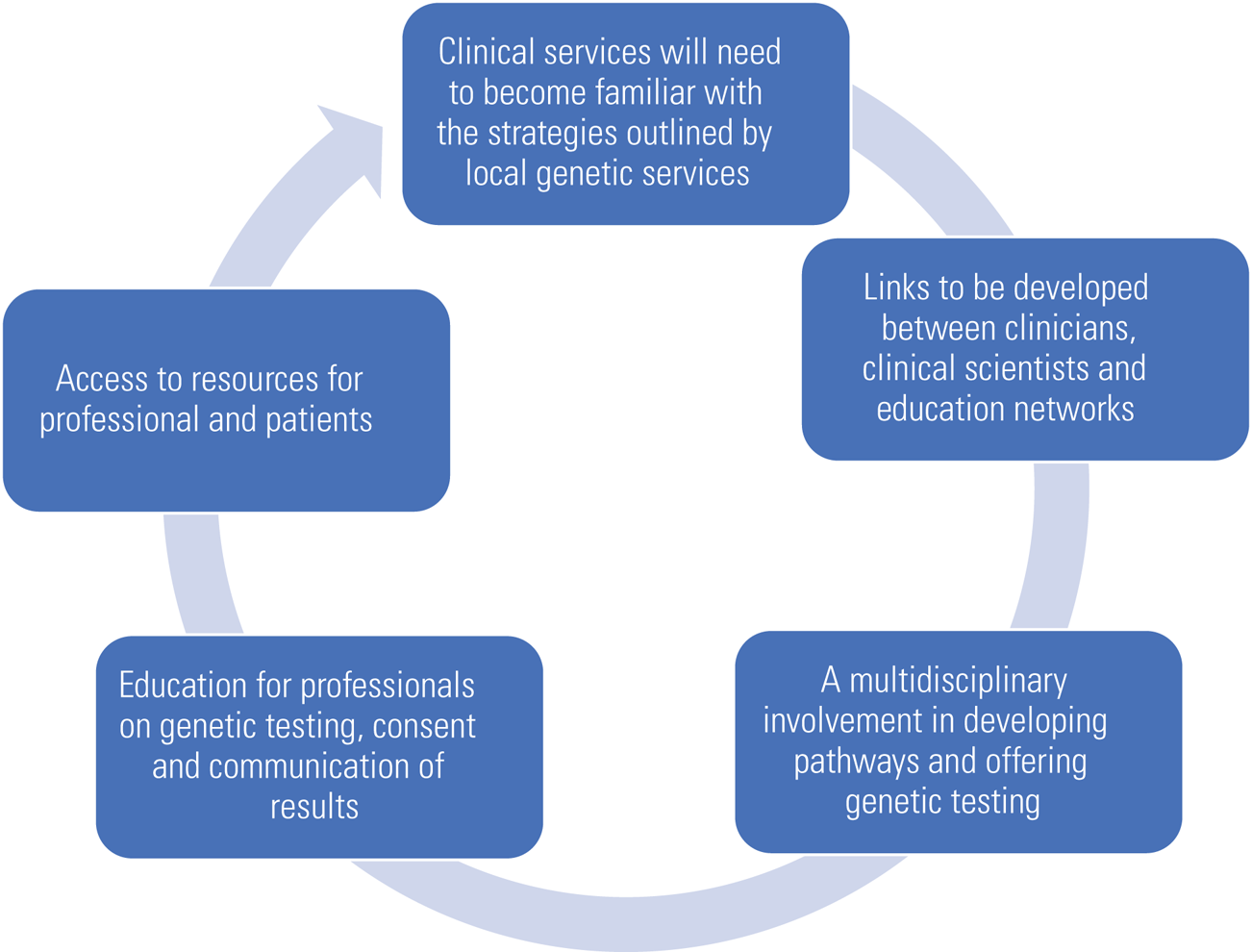
FIG 1 Preparing for change (author's own diagram).
Evolution of the genetic testing techniques
Cytogenetics has been a key part of biology since the discovery of chromosomes in 1842. Since the ability to detect chromosomal changes using karyotyping, genetic testing has advanced to be able to detect smaller genetic variations. As research began to correlate known clinical syndromes with specific variations in genetic material, fluorescence in situ hybridisation (FISH) probes were developed to identify these differences. However, this method relied on a high level of clinical suspicion and was limited to looking for variation in one region only.
In more recent years, both earlier methods have largely been replaced by microarray-based comparative genomic hybridisation (array CGH). This method has provided an alternative way of genome-wide screening for copy number variants (CNVs) and can detect rearrangements of at least 100 000 to 200 1000 base pairs in size as part of a chromosome in the form of deletions and/or duplications of any locus represented on an array in an automated way; it achieves this by using two genomes, a test and a control, which are differentially labelled. It provides information on how much genetic material is present at each genetic locus, but not where it is in the DNA strand. Microarrays are often used as an initial test in children and adults with multiple congenital abnormalities and/or developmental delay or epilepsy. Its limitation is that it does not detect low-level mosaicism (below 10%; Hoang Reference Hoang, Ahn and Mann2010), single-gene variants, balanced translocations, imprinting disorders due to methylation abnormalities or fragile-X syndrome (Gray Reference Gray, Kallioniemi and Kallioniemi1992; Miller Reference Miller, Adam and Aradhya2010).
Since the wide use of these next-generation sequencing technologies, greater diagnostic yield with rapid diagnosis, ultra-high throughput, scalability and speed have been achieved. It is a massively parallel DNA sequencing technology that offers diagnosis of conditions with high genetic heterogeneity in the form of single-gene testing, gene-panel testing, whole-exome (WES) and whole-genome sequencing (WGS) (Box 3). Through WES and WGS, novel genetic conditions can be identified that were not previously linked to a clinical phenotype (Newman Reference Newman and Black2014). The limitations of WGS include its inability to detect deletions or duplications, structural chromosomal rearrangements, ring chromosomes, triplet repeats and epigenetic variations.
BOX 3 Next-generation DNA sequencing technologies
Gene-panel testing
Gene-panel testing is used for detection of genetic changes in a group of genes that are associated with a condition or similar phenotype. It focuses on a predetermined group of genes (e.g. genes associated with epilepsy).
Whole-exome sequencing (WES)
WES involves sequencing only the specific sequences of DNA within the genome that code for proteins (exons, which make up 1% of our total DNA) of all known genes. A specific ‘virtual panel’ of genes of interest can also be selected for initial analysis and the analysis can be extended to include additional genes if required.
Whole-genome sequencing (WGS)
WGS refers to sequencing the entire genome (>3 billion base pairs) of an individual, including coding and non-coding regions. Analysis can also be performed with a ‘virtual panel’ of genes of interest and be broadened to look for changes in genes that have not previously been associated with a clinical phenotype.
Why test?
Genetic testing can potentially offer a definitive diagnosis and inform clinicians and patients in deciding appropriate treatment options or access to necessary support services. It may also help to reduce the number of sometimes invasive investigative procedures and maltreatment with complex unnecessary therapies. Certainty about a diagnosis can provide relief following years of uncertainty.
By performing genetic testing in the context of intellectual disability, epilepsy and ASD in children and adults, enhancement of prevention and patient care can be achieved through the identification of previously unrecognised comorbidities and active surveillance and early intervention before symptoms develop. It can provide patients, parents/carers and the clinical team with information about likely clinical problems and complications as well as long-term prognosis, enabling patients and their families to access special education, social care and employment opportunities. Genetic findings can enable prediction of recurrence risks for the offspring of the affected individual and their relatives and allow discussion in the context of family planning, eligibility for pre-implantation genetic diagnosis in the future and prenatal testing in pregnancy. A diagnosis could also help patients to access research trials and to get in touch with other similarly affected individuals or families (Vasudevan Reference Vasudevan2017; Savatt Reference Savatt and Myers2021). Specificity in diagnosis can allow robust patient groups to be found for research, and with advances in genomic medicine there may be potential for gene-specific interventions in future.
Which genetic test to use?
Moving from the chromosome band (cytogenetics) to the DNA base pair (sequencing) can provide greater accuracy, time efficiency and cost-effectiveness (Fig. 2). The cytogenetic tests commonly used to identify genetic changes that predispose to intellectual disability, ASD and epilepsy are the chromosomal microarrays (array CGH and single-nucleotide polymorphism (SNP) array) and polymerase chain reaction (PCR) (for fragile-X syndrome). Karyotyping is currently used rarely and only to detect large structural chromosomal rearrangements and mosaicism.

FIG 2 Genetic tests. FISH, fluorescence in situ hybridisation (Health Education England, Genomics Education Programme image library).
However, the key in deciding the most effective testing method still lies in the careful clinical phenotyping of each patient: the detailed clinical examination, the family history and other relevant investigations that add information to the diagnostic puzzle. For adults, changes in diagnostic criteria for neurodevelopment disorders and epilepsy syndromes that have taken place over the years can result in poor clarity in definition of clinical phenotype, which may need to be carefully reviewed. A combination of tests is used in most cases to aid the genetic diagnosis.
Results from genetic testing may be difficult to interpret. Newer techniques recommend comparison of genetic variation with parental samples (exome trio analysis), which may not be possible in some adults or in individuals who are fostered or adopted. The most recent pilot study of data from the 100 000 Genome Project looking at rare disorders indicated that the diagnostic yield for whole-genome sequencing was highest when information was also available for both parents (trio analysis) and for families with a larger pedigree. For example, for those with intellectual disability, diagnostic yield was 25% in singletons, 41.7% for duos and 45.1% for trios (The 100 000 Genomes Project Pilot Investigators 2021).
Variations may be found in genes that have not previously been described in medical literature or that do not explain the individual's clinical phenotype. This is referred to as a variant of uncertain significance (VUS). Careful interpretation of genetic findings in relation to clinical features and research findings is required to avoid inaccurate over-interpretation of variants. Incomplete penetrance of some autosomal dominant disorders can also confound diagnostic clarity.
It is important to be aware that some syndromes require additional specific testing, as they are not readily identified using current common methods (Table 1). This includes disorders of mitochondrial DNA (mtDNA), imprinting disorders (e.g. Angelman syndrome, for which methylation testing is used) and fragile-X syndrome.
TABLE 1 Comparison of current methods of genetic testing

Robust guidance has been created to ensure standardised ways of interpreting genetic findings, which is essential to avoid inaccurate diagnosis that might lead to incorrect clinical advice regarding pre-diagnostic testing and intervention. Genetic variants are classified according to guidelines agreed through the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (Box 4).
BOX 4 Classifications of genetic variants
Benign variant
Not thought to be associated with disease. Such variants are often present in phenotypically normal individuals or in the general population.
Likely benign variant
Considerable evidence to suggest that the variant is not associated with disease but additional evidence could further clarify this.
Variant of uncertain significance
There is not enough information to determine whether the variant is pathogenic or benign. Parental studies might be used to provide additional information to clarify pathogenicity.
Likely pathogenic variant
Considerable evidence to suggest that the variant is associated with disease but additional evidence could further clarify the variant's pathogenicity.
Pathogenic
Thought to be associated with disease. The variant is known to be associated with a rare condition and to explain the patient's phenotype.
Neurodevelopmental disorders and diagnostic yield from genetic testing
Medical investigations have a higher diagnostic yield in children with severe developmental delay (developmental quotient 4 s.d. or more below the mean on formal testing) than in the larger group of children with mild or moderate delay (developmental quotient 2–4 s.d. below the mean on formal testing).
Neurodevelopmental disorders may arise from genetic variation that affects brain development and function. Genetic variation can occur within a chromosomal change, a copy number variant or a single nucleotide change. Epigenetic processes also affect how and when genes are transcribed into proteins. These processes regulate the changes required at different stages of development. DNA methylation and chromatin modification are two global mechanisms that regulate gene expression (Ng Reference Ng and Bird1999). The human genome is tightly packaged with chromatin in the nucleus of the cell, with DNA sequences wrapped around histone proteins. Chromatin modification takes place via enzyme reactions to allow specific regions of DNA to be accessed. Methylation is a chemical reaction that enables sequences of DNA to be ‘silenced’ to regulate expression.
Genes that influence early brain development and neuronal communication (synaptic function) (Satterstrom Reference Satterstrom, Kosmicki and Wang2020) and those that regulate the expression of other genes are highly implicated in neurodevelopmental disorders (Sanders Reference Sanders, He and Willsey2015).
Intellectual disability
Chromosomal microarray is considered the first-line diagnostic genetic test to look for copy number variants in all individuals with global developmental delay/intellectual disability (Moeschler Reference Moeschler and Shevell2014). Diagnostic yields from microarray testing have been found to be up to 13% in adults with intellectual disability and comorbid psychiatric disorders (Thygesen Reference Thygesen, Wolfe and McQuillin2018).
Fragile-X syndrome remains a common cause of intellectual disability and ASD, and even epilepsy in some cases, and should be offered in addition to microarray testing to those with a positive family history of intellectual disability and autism.
Fragile-X syndrome is caused by a short tandem repeat (STR) on the X chromosome, as a result of transcriptional silencing of the FMR1 gene, leading to the absence of the FMR1 protein, which plays a key role in prenatal and postnatal brain development and has a critical function in the establishment and maintenance of neuronal synapses. The level of intellectual disability relates to the length of the tandem repeat. Males typically present with moderate to severe developmental delay/intellectual disability and females often have milder disability. Individuals with fragile-X may experience early cognitive decline and tremor (Sauna-Aho Reference Sauna-Aho, Bjelogrlic-Laakso and Rautava2020).
The diagnostic yield of fragile-X syndrome in global developmental delay/intellectual disability and/or ASD varies widely with study design, with yields reported between 0.5 and 6% (Peprah Reference Peprah2012). A large-scale study has suggested that fragile-X is substantially underdiagnosed in the general population owing to clinical heterogeneity (Movoghar Reference Movoghar, Page and Brilliant2021).
With the introduction of next-generation sequencing, novel single-gene disorders are being discovered which contribute to intellectual disability (Järvelä Reference Järvelä, Määttä and Acharya2021), with up to 75% of diagnoses occurring from de novo variants. A de novo variant is a variant that is not present in either biological parent. Gene panels have been created to combine the known common genes for intellectual disability that can be applied to the genome sequence.
Large cohort studies are comparing the diagnostic yield of array CGH and next-generation sequencing for genetic disorders. One such (Martinez-Granero Reference Martinez-Granero, Blanco-Kelly and Sanchez-Jimeno2021) highlights the importance of gathering as much clinical information as possible to help guide genetic testing and interpretation of results (Fig. 3).

FIG 3 Comparison of the yield of genetic diagnosis from microarray-based comparative genomic hybridisation (aCGH) and from next-generation sequencing (NGS) (data from Martinez-Granero et al, Reference Martinez-Granero, Blanco-Kelly and Sanchez-Jimeno2021).
Autism spectrum disorder
Autism spectrum disorder (ASD) is the diagnostic term used to describe individuals with impairment in social communication and restricted or repetitive patterns of behaviour.
Chromosomal microarray is considered the first-line diagnostic genetic test in all individuals with ASD. Fragile-X testing should also be offered to those with a positive family history of intellectual disability and ASD.
A strong genetic component to ASD has been consistently shown in family and twin studies, with estimated heritability ranging from 40 to 90% (Gaugler Reference Gaugler, Klei and Sanders2014). Research involving a longitudinal study from the Baby Siblings Research Consortium found that 35.8% of siblings were diagnosed with ASD and 18.8% were developing atypically (D'Abate Reference D'Abate, Walker and Yuen2019).
The genetic basis of ASD is complex, with polygenic inheritance and potential environmental factors contributing to phenotypic variation. Up to 40% of individuals with ASD are now diagnosed with genetic syndromes or have chromosomal abnormalities such as small DNA deletions, duplications, single-gene conditions or gene variants and metabolic disturbance with mitochondrial dysfunction (Genovese Reference Genovese and Butler2020).
In a small study of adults with ASD, array CGH revealed that 21% of participants had a pathogenic copy number variant, including one with fragile-X syndrome (Stobbe Reference Stobbe, Liu and Wu2014). This highlights the importance of genetic re-evaluation in such populations.
Strong associations with de novo copy number variants have been replicated across a number of large-scale studies with risk DNA loci for ASD confirmed at 1q21.1, 3q29, 7q11.23, 16p11.2, 15q11.2-13 and 22q11.2 (Sanders Reference Sanders, He and Willsey2015).
The Autism Sequencing Consortium has created a multisite collaboration to recruit and combine large cohorts to understand more about the genetic contribution to ASD (Buxbaum Reference Buxbaum, Daly and Devlin2012).
The yield for whole-genome sequencing is low in those diagnosed with ASD alone but increases when the disorder is accompanied by a history of delayed motor development, intellectual disability and/or seizures (Sanders Reference Sanders, He and Willsey2015; Martinez-Granero Reference Martinez-Granero, Blanco-Kelly and Sanchez-Jimeno2021). This highlights the importance of gathering information on early developmental milestones. Specific clinical features may suggest a specific genetic condition, for example macrocephaly and PTEN gene variation (Kaymakcalan Reference Kaymakcalan, Kaya and Binici N2021). A deviation in head circumference of more than 1 s.d. in either direction is associated with a higher diagnostic yield (Chaste Reference Chaste, Klei and Sanders2013).
Most research to date has focused on areas of the genome known to code for specific proteins. More recently, research has focused on de novo variants in non-coding areas of the genome. These non-coding variants, known as enhancers, regulate the transcription of other genes and have been found to be enriched in individuals with autism (Turner Reference Turner, Coe and Dickel2017).
Epilepsy
Epilepsy affects approximately 0.6 to 0.8% of the general population; a large proportion of cases are of unknown aetiology, with wide variation and a complex mode of inheritance (polygenic and complex genetic aetiology).
Genetic tests have played a significant role in the diagnostic odyssey of epilepsy by identifying variants of likely pathogenic or definitive clinical significance in developmental and epileptic encephalopathies, with a shift towards using genome sequencing in unsolved cases (Happ Reference Happ and Carvill2020). It is estimated that more than half of all epilepsies have a genetic basis (Pal Reference Pal, Pong and Chung2010). Genetic factors are also thought to contribute to intractable epilepsy and status epilepticus (Corey Reference Corey, Pellock and Boggs1998).
It is important to characterise the epilepsy phenotype and consider age at onset, with a detailed gestational, birth, developmental and family history, when considering genetic testing in adults with epilepsy, as different testing strategies are followed (e.g. for early-onset epilepsies).
A comparison of genetic tests in epilepsy (Sánchez Fernández Reference Sánchez Fernández, Loddenkemper and Gaínza-Lein2019) reported diagnostic yields and cost-effectiveness as follows:
• 0.06–0.12 for array CGH (cheap)
• 0.18–0.29 for epilepsy gene panel (cost-effective)
• 0.33–0.57 for whole-exome sequencing (expensive).
Microarray testing should be ordered in patients with epilepsy with intellectual disability/global developmental delay/ASD as this is currently the most cost-effective and clinically useful test in this group. Next-generation sequencing should be considered based on specific clinical features, electroencephalogram (EEG) patterns and magnetic resonance imaging (MRI) brain findings (Nashef Reference Nashef, Singh and Moran2019).
Genes associated with epilepsy are divided into three main categories: epilepsy genes, neurodevelopment-associated epilepsy genes and epilepsy-related genes (Box 5).
BOX 5 Gene categories associated with epilepsy
Epilepsy genes
The term is used when epilepsy is the exclusive outcome of the mutation or when part of a group of unrelated symptoms and mutations in these genes can cause pure or relatively pure epilepsies or syndromes with epilepsy as the core symptom (Wang Reference Wang, Lin and Liu2017). The spectrum of epilepsy caused by gene mutations covers multiple epileptic phenotypes, ranging from the mild form of benign familial infantile seizures to the extremely severe form of early infantile epileptic encephalopathy.
Neurodevelopment-associated epilepsy genes
Many genes have been classified as neurodevelopment-associated epilepsy genes. Mutations in these genes produce gross neurodevelopmental malformations and epilepsy, which may vary in severity.
Epilepsy-related genes
Potential epilepsy-associated genes that warrant further verification in most cases.
Many ‘idiopathic’ epilepsies have now been found to be caused by genetic abnormalities, and mutations have been identified in several genes, including GABRG2, GABRB3, CACNA1H and GABRA1 (Wang Reference Wang, Lin and Liu2017; Dunn Reference Dunn, Albury and Maksemous2018).
Genetic testing is carried out to help with the diagnosis and prognosis and to anticipate difficulties and it can directly guide intervention with anti-epileptic medication, ketogenic diet or surgery, depending on the variant identified: for example, anti-epileptic medication for sodium channel mutations involving the SCN1A and SCN2A genes (Musto Reference Musto, Gardella and Møller2020; Ademuwagun Reference Ademuwagun, Rotimi and Syrbe2021), pyridoxine for pyridoxine-dependent epilepsy involving the ALDH7A1 gene (Plecko Reference Plecko, Paul and Paschke2006) and ketogenic diet for GLUT1 deficiency syndrome involving the SLC2A1 gene (Klepper Reference Klepper, Akman and Armeno2020) – while valproic acid-induced hepatotoxicity in a mitochondrial disorder such as POLG gene mutations might be avoided (Saneto Reference Saneto, Lee and Koenig2010).
The challenges of diagnosing epileptic syndromes lie in the genetic heterogeneity and the fact that one gene variant can manifest multiple clinical epilepsy phenotypes (e.g. SCN1A, KCNQ2, GABRA1) (Meng Reference Meng, Xu and Yu2015). There are some genetic conditions that may be missed by microarray and next-generation sequencing, for example ring chromosome 20 in individuals with non-convulsive status, which requires karyotyping (Peron Reference Peron, Catusi and Recalcati2020).
Practical pathway and tool kit for genetic testing
Which genetic test should I order?
• All individuals with intellectual disability, autism or epilepsy should be offered genetic testing with microarray to look for copy number variants.
• Consider fragile-X testing in those with positive family history of intellectual disability and autism spectrum disorder in both males and females (fragile-X is unlikely in individuals with microcephaly).
• Consent is not needed to store DNA – so if patients are having a blood test for another reason there is nothing to be lost by requesting a sample which can be stored for many years. Saliva can be used but is not quite as good and should be discussed with the laboratory before sending.
When should a referral be made to the clinical genetics team for an individual with intellectual disability?
• In the absence of a genetic diagnosis from microarray and fragile-X testing, consider referral to clinical genetics services for next-generation sequencing.
• When referring, carefully consider whether there are any specific features that may help with the interpretation of genetic findings: severe intellectual impairment, overgrowth, micro/macrocephaly, dysmorphic features, congenital anomalies.
• If uncertain it is always helpful to talk with the local genetics team or regional genomics laboratory hub (Fig. 4).
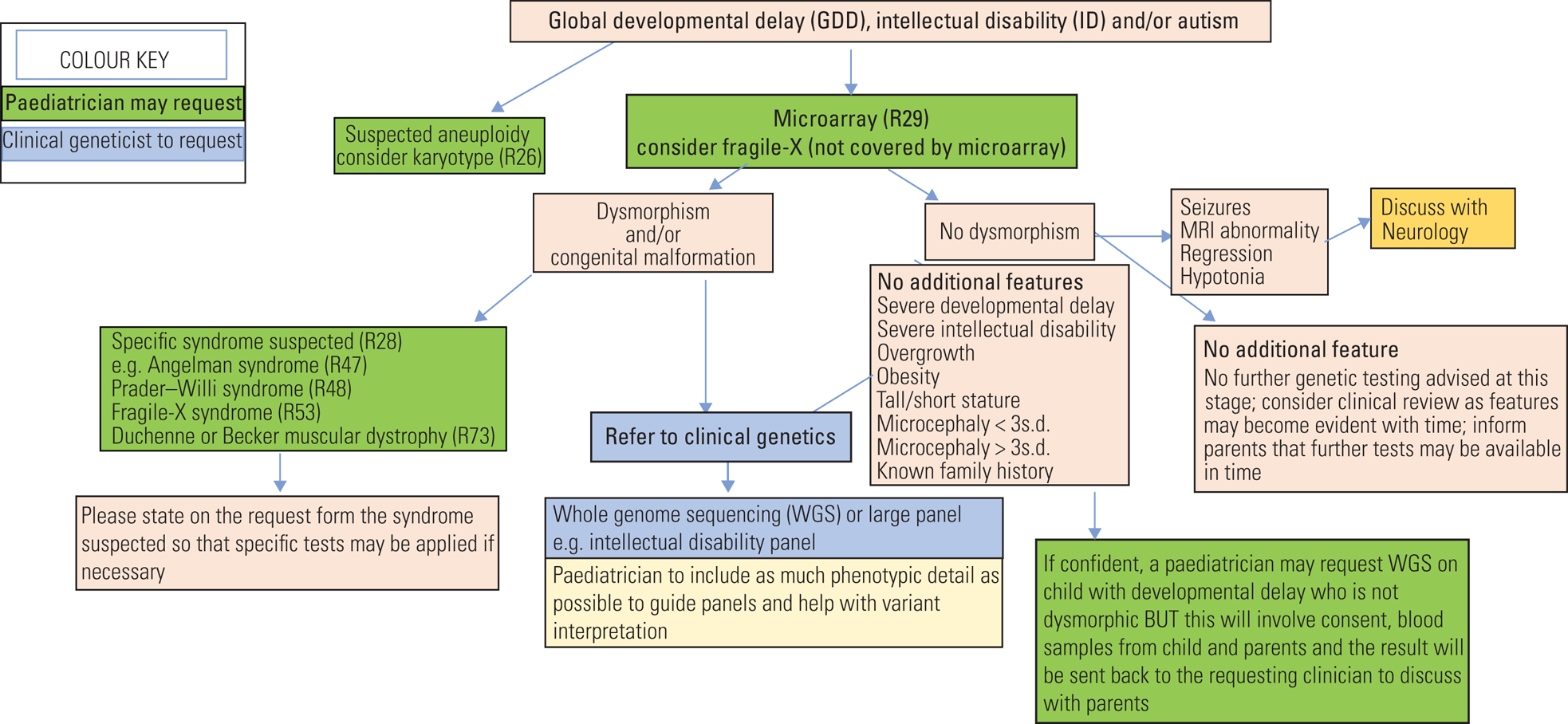
FIG 4 Flow chart guidance for genetic investigations of global developmental delay, intellectual disability and autism spectrum disorder.
Where can I find patient information on genetic testing?
• General information from the NHS: www.nhs.uk/conditions/genetic-and-genomic-testing/
• Online information, leaflets, videos and animations (including easy-read and translated versions), e.g.:
◦ the North London genomics laboratory hub: www.norththamesglh.nhs.uk/patients-and-public/
◦ Great Ormond Street Hospital for Children (www.gosh.nhs.uk): search under ‘clinical genetics support and information’ for, e.g. an animated resource available in several languages for young people and individuals with intellectual disability.
Where can I find information about specific gene findings?
Resources aimed at health professionals give key facts about each condition, plus detailed information on clinical features, diagnosis, management and treatment:
• Genomics Education Programme genetic conditions factsheets: www.genomicseducation.hee.nhs.uk/doc-type/genetic-conditions
• Unique: www.rarechromo.org
• Online Mendelian Inheritance in Man (OMIM): omim.org
• Genetic Alliance UK: geneticalliance.org.uk
• PubMed Central: www.ncbi.nlm.nih.gov/pmc
• MedlinePlus: medlineplus.gov/genetics
Additional resources:
• www.undiagnosed.org.uk (for SWAN – syndromes without a name – where testing has failed to identify a genetic cause in an individual thought to have a genetic condition).
Author contributions
E.C. and M.K. jointly contributed to the design and content of the work, including drafting and revising it critically for important intellectual content and final approval of the version to be published. E.C. and M.K. agree to be accountable for ensuring that questions related to the accuracy and integrity of any part of the work are appropriately investigated and resolved. E.C. and M.K. are very grateful for the support of contributors to this article from the North Thames Regional Genetics Service Great Ormond Street Hospital: Dr Emma Wakeling, Consultant in Clinical Genetics; Dr Elisabeth Rosser, Consultant in Clinical Genetics; and Dr Lara Menzies, Consultant in Clinical Genetics. E.C. and M.K. would like to acknowledge the learning opportunities received through their clinical fellow roles with the North Thames Laboratory Hub within the Genomics Medicine Service Alliance.
Funding
This research received no specific grant from any funding agency, commercial or not-for-profit sectors.
Declaration of interest
None.
MCQs
Select the single best option for each question stem
1 The following genetic test can detect single base pair changes:
a karyotype
b microarray
c fragile-X testing
d whole-genome sequencing
e FISH probe.
2 Evidence shows that the probability of finding a rare single-gene disorder is increased with:
a presence of a specific learning disorder
b male gender
c speech delay
d microarray testing
e exome trio analysis.
3 A genetic diagnosis can help with:
a personalised medical care
b understanding coexisting conditions and offering appropriate surveillance
c understanding recurrence risks and prenatal testing
d long-term prognosis to access support networks
e all of the above.
4 The following is true of fragile-X syndrome:
a it can only occur in males
b it is associated with microcephaly
c it is thought to be substantially underdiagnosed in the general population
d a shorter tandem repeat is associated with a higher level of intellectual disability
e it never occurs in females.
5 Genetic investigations for intellectual disability, epilepsy and autism might include:
a first-line testing with microarray
b considering fragile-X testing in those with intellectual disability and autism
c whole-genome sequencing
d referral to clinical genetics if there are no pathogenic findings on microarray or fragile-X testing
e all of the above.
MCQ answers
1 d 2 e 3 e 4 c 5 e






eLetters
No eLetters have been published for this article.