



Implications
Long non-coding RNAs are common transcripts existing in genomes, including those of livestock. An individual transcriptome contains more long non-coding RNAs than messenger RNA molecules. Our literature review shows significant impact of long non-coding RNAs on a variety of phenotypes relevant to livestock production and welfare, which is exhibited by the role of long non-coding RNAs as modifiers of the expression of protein-coding genes. In humans, long non-coding RNAs exist which act as disease biomarkers what can also be anticipated for livestock. The better understanding of individual- and tissue-specific variability in long non-coding RNAs expression is important for more precise exploitation of genetic variation of livestock phenotypes.
Introduction
Beginning from the discovery of transfer RNA (tRNA) and ribosomal RNA (rRNA) in the 1950s, non-coding RNAs (ncRNAs) with biological roles have been known for some 60 years (Palazzo and Lee Reference Palazzo and Lee2015). Recent advances in next-generation sequencing methods have been proving new possibilities of investigating the full set of RNA molecules in genomes and have led to the increase in the number of studies carried out on RNA. Specifically, RNA-seq is a technique used for a whole transcriptome sequencing which, together with computational methods, allows for transcriptome reconstructing and the quantification of gene expression. The method overcomes shortcomings of microarray technology by offering a more comprehensive coverage of whole transcriptomes, and it is not limited to known sequences (Uchida et al., Reference Uchida2017). Therefore, RNA-seq offers a remarkable opportunity to genome-wide annotation and characterization of long non-coding RNAs (lncRNAs) (Xiao et al., Reference Xiao, Hu and Yin2018). According to recent knowledge, less than 2% of the genome codes for proteins, but ‘the majority of its bases can be found in primary transcripts’ – a phenomenon termed the pervasive transcription, which was first reported by the ENCODE Project Consortium (Reference Birney, Stamatoyannopoulos, Dutta, Guigó, Gingeras, Margulies, Weng, Snyder, Dermitzakis, Thurman, Kuehn, Taylor, Neph, Koch, Asthana, Malhotra, Adzhubei, Greenbaum, Andrews, Flicek, Boyle, Cao, Carter, Clelland, Davis, Day, Dhami, Dillon, Dorschner, Fiegler, Giresi, Goldy, Hawrylycz, Haydock, Humbert, James, Johnson, Johnson, Frum, Rosenzweig, Karnani, Lee, Lefebvre, Navas, Neri, Parker, Sabo, Sandstrom, Shafer, Vetrie, Weaver, Wilcox, Yu, Collins, Dekker, Lieb, Tullius, Crawford, Sunyaev, Noble, Dunham, Denoeud, Reymond, Kapranov, Rozowsky, Zheng, Castelo, Frankish, Harrow, Ghosh, Sandelin, Hofacker, Baertsch, Keefe, Dike, Cheng, Hirsch, Sekinger, Lagarde, Abril, Shahab, Flamm, Fried, Hackermüller, Hertel, Lindemeyer, Missal, Tanzer, Washietl, Korbel, Emanuelsson, Pedersen, Holroyd, Taylor, Swarbreck, Matthews, Dickson, Thomas, Weirauch, Gilbert, Drenkow, Bell, Zhao, Srinivasan, Sung, Ooi, Chiu, Foissac, Alioto, Brent, Pachter, Tress, Valencia, Choo, Choo, Ucla, Manzano, Wyss, Cheung, Clark, Brown, Ganesh, Patel, Tammana, Chrast, Henrichsen, Kai, Kawai, Nagalakshmi, Wu, Lian, Lian, Newburger, Zhang, Bickel, Mattick, Carninci, Hayashizaki, Weissman, Hubbard, Myers, Rogers, Stadler, Lowe, Wei, Ruan, Struhl, Gerstein, Antonarakis, Fu, Green, Karaöz, Siepel, Taylor, Liefer, Wetterstrand, Good, Feingold, Guyer, Cooper, Asimenos, Dewey, Hou, Nikolaev, Montoya-Burgos, Löytynoja, Whelan, Pardi, Massingham, Huang, Zhang, Holmes, Mullikin, Ureta-Vidal, Paten, Seringhaus, Church, Rosenbloom, Kent, Stone, Batzoglou, Goldman, Hardison, Haussler, Miller, Sidow, Trinklein, Zhang, Barrera, Stuart, King, Ameur, Enroth, Bieda, Kim, Bhinge, Jiang, Liu, Yao, Vega, Lee, Ng, Shahab, Yang, Moqtaderi, Zhu, Xu, Squazzo, Oberley, Inman, Singer, Richmond, Munn, Rada-Iglesias, Wallerman, Komorowski, Fowler, Couttet, Bruce, Dovey, Ellis, Langford, Nix, Euskirchen, Hartman, Urban, Kraus, Van Calcar, Heintzman, Kim, Wang, Qu, Hon, Luna, Glass, Rosenfeld, Aldred, Cooper, Halees, Lin, Shulha, Zhang, Xu, Haidar, Yu, Ruan, Iyer, Green, Wadelius, Farnham, Ren, Harte, Hinrichs, Trumbower, Clawson, Hillman-Jackson, Zweig, Smith, Thakkapallayil, Barber, Kuhn, Karolchik, Armengol, Bird, de Bakker, Kern, Lopez-Bigas, Martin, Stranger, Woodroffe, Davydov, Dimas, Eyras, Hallgrímsdóttir, Huppert, Zody, Abecasis, Estivill, Bouffard, Guan, Hansen, Idol, Maduro, Maskeri, McDowell, Park, Thomas, Young, Blakesley, Muzny, Sodergren, Wheeler, Worley, Jiang, Weinstock, Gibbs, Graves, Fulton, Mardis, Wilson, Clamp, Cuff, Gnerre, Jaffe, Chang, Lindblad-Toh, Lander, Koriabine, Nefedov, Osoegawa, Yoshinaga, Zhu and Jong2007). Recently, Lee et al. (Reference Lee, Park, Lee, Lee and Kim2018) stated that only 1~2% of the genome has a protein-coding potential, while the reminder forms ncRNA molecules. Because of typically low expression levels (comparing to protein-coding transcripts), they are described as ‘transcription noise’ (Ma et al., Reference Ma, Bajic and Zhang2013).
Non-coding RNA is classified into two groups: short and long non-coding RNA (Nie et al., Reference Nie, Wu, Hsu, Chang, Labaff, Li, Wang, Hsu and Hung2012). Transcripts shorter than 200 nucleotides are termed small non-coding RNA and include Piwi-interacting RNA, small interfering RNA, microRNA (miRNA), rRNA, tRNA, small nucleolar RNA and small nuclear RNA (Storz, Reference Storz2002). Transcripts longer than 200 nucleotides are classified as lncRNAs among which lncRNAs that located in-between genes are termed long intergenic non-coding RNA (lincRNA) (Wang and Chang, Reference Wang and Chang2011; Zheng et al., Reference Zheng, Ning, Zhao, Feng, Jin, Zhou, Yu and Liu2018).
In livestock, genome-wide association studies based on single-nucleotide polymorphisms (SNPs) led to the identification of many mutations causal for the phenotypes of commercial interest, still most of the significant SNPs fall into genomic regions not covered by genic DNA (Goddard et al., Reference Goddard, Kemper, MacLeod, Chamberlain and Hayes2016; www.animalgenome.org/QTLdb/). However, despite this large number of significant SNPs identified, their joint effects do not account for all of the phenotypic variations observed in traits routinely measured in livestock – the phenomenon called ‘missing heritability’ first introduced into human genetics by Manolio et al. (Reference Manolio, Collins, Cox, Goldstein, Hindorff, Hunter, McCarthy, Ramos, Cardon, Chakravarti, Cho, Guttmacher, Kong, Kruglyak, Mardis, Rotimi, Slatkin, Valle, Whittemore, Boehnke, Clark, Eichler, Gibson, Haines, Mackay, McCarroll and Visscher2009). Since lncRNA may be one of the potential causes of missing heritability, it is of interest for livestock genomics. Therefore, the aim of our study was to characterize the current state of knowledge on lncRNA in three major species of farm animals: Bos taurus, Sus scrofa and Gallus gallus.
Long non-coding RNA detection workflow
Given a raw RNA-seq data, a typical workflow for the identification and annotation of lncRNAs is composed of two major parts – a part which is common to processing all RNA-seq data and a part dedicated to lncRNA.
In the common part, the first step (1a) involves generation of a control report for sequence quality, which is typically done using the FastQC software (Andrews et al., Reference Andrews2010). In the second step (2a), raw sequence reads are pre-processed by filtering out contaminations from sequencing adapters and by removing or trimming low-quality reads. The minimum threshold for read quality score is usually set to 20 (Wu et al., Reference Wu, Liu, Guo, Cheng, Ren, Chen, Li, Duan, Sun and Yang2018). The Trimmomatic software (Bolger et al., Reference Bolger, Lohse and Usadel2014) is a popular tool for pre-processing of raw RNA-seq data. In the next step (3a), cleaned sequences are mapped to the reference genome with the most commonly used software tools being: Tophat (Trapnell et al., Reference Trapnell, Pachter and Salzberg2009), Tophat2 (Kim et al., Reference Kim, Pertea, Trapnell, Pimentel, Kelley and Salzberg2013), Bowtie2 (Langmead and Salzberg Reference Langmead and Salzberg2012) or HISAT2 (Kim et al., Reference Kim, Langmead and Salzberg2015). Finally in the last step (4a), assembling of the uniquely mapped sequence reads into transcripts is most often done by Cufflinks (Trapnell et al., Reference Trapnell, Williams, Pertea, Mortazavi, Kwan, van Baren and Pachter2010) or StringTie (Pertea et al., Reference Pertea, Pertea, Antonescu, Chang, Mendell and Salzberg2015).
In order to proceed with the analysis specific to lncRNA, several transcript filtration steps are to be pre-imposed on the annotated RNA-seq data. The first step (1b) is to identify only novel transcripts, which do not correspond to the protein-coding part of the genome and do not represent previously annotated lncRNAs. It is most often performed using the Cuffcompare function of Cufflinks, which classifies all the available assembled transcripts based on their annotation to the pre-specified reference genome in the GFF format. Transcripts classified as ‘A transfrag falling entirely within a reference intron’, ‘Unknown, intergenic transcript’ or ‘Exonic overlap with reference on the opposite strand’ are then selected for downstream analysis. The genomic annotation of lncRNA in livestock is still very scarce as compared to human or mouse genomes; therefore, for livestock species, a much larger number of novel lncRNA transcripts is expected than for humans. Such potential candidate sequences for lncRNA are then subjected to several filtering steps (2b), which can differ depending on the analytic approach. The most important filtering steps comprise (i) filtering by length – removing transcripts shorter than 200 bp and transcripts longer than 10 000 bp containing a single exon, (ii) filtering by sequence content – removing transcripts overlapping with repeat or low complexity regions defined in the reference genome assembly (note, that for livestock genomes, this information is still limited), (iii) filtering by expression level – removing extremely high and extremely low expressed transcripts using the FPKM measure (fragments per kilo base of transcript per million mapped reads) to quantify the expression level; removal thresholds can either be arbitrarily chosen or estimated dynamically from the available data and (iv) filtering by protein-coding potential – various approaches involving removing transcripts containing known protein-coding domains using, for example, Transeq (El-Gebali et al., Reference El-Gebali, Mistry, Bateman, Eddy, Luciani, Potter, Qureshi, Richardson, Salazar, Smart, Sonnhammer, Hirsh, Paladin, Piovesan, Tosatto and Rober Finn2019) or HMMER (Eddy et al., Reference Eddy2011; Finn et al., Reference Finn, Clements and Eddy2011) software; removing transcripts with a significant hit in the Pfam database using, for example, PfamScan software (Bateman et al., Reference Bateman, Birney, Cerruti, Durbin, Etwiller, Eddy, Griffin-Jones, Howe, Marshall and Sonnhammer2002; Finn et al., Reference Finn, Bateman, Clements, Coggill, Eberhardt, Eddy, Heger, Hetherington, Holm, Mistry and Sonnhammer2014); removing transcripts, which products show similarity to known proteins from the RefSeq non-redundant protein database or the UniRef90 database (Suzek et al., Reference Suzek, Wang, Huang, McGarvey and Wu2015) using BLASTX (Altschul et al., Reference Altschul, Gish, Miller, Myers and Lipman1990); removing transcripts based on their protein-coding potential level estimated, for example, by Coding Potential Calculator (CPC) (Kong et al., Reference Kong, Zhang, Ye, Liu, Zhao, Wei and Gao2007), Coding Potential Assessment Tool (CPAT) (Wang et al., Reference Wang, Park, Dasari, Wang and Kocher2013), Coding Non-Coding Index (CNCI) software (Sun et al., Reference Sun, Luo, Bu, Zhao, Yu, Zhang, Liu, Chen and Zhao2013) or predictor of Long non-coding RNAs and mEssenger RNAs based on an improved K-mer scheme (PLEK) (Li et al., Reference Li, Zhang and Zhou2014). The next step (3b) comprises merging of the RNA-seq data corresponding to novel lncRNAs, defined by the above workflow, and the known lncRNAs with positions defined in databases such as the ALDB (Li et al., Reference Li, Ma, Zeng, Wang, Geng, Yang and Cui2015) or the NONCODE. Note that various authors apply different thresholds for the percent of sequence identity and the percent of length of the aligned sequence to call lncRNAs. The final downstream analysis (4b) of the combined data sets depends on the experimental hypothesis and on the underlying experimental design. Most typically, it involves (i) the comparison of lncRNA expression levels between experimental conditions using, for example, DESeq2 software (Love et al., Reference Love, Huber and Anders2014), (ii) identification of target genes of differentially expressed lncRNAs, which can either be done by a dedicated software, for example, LncTar (Li et al., Reference Li, Ma, Zeng, Wang, Geng, Yang and Cui2015), by considering the physical proximity between a lncRNA and a protein-coding gene, or by considering high correlations between the expression level of a lncRNA and an messenger RNA (mRNA), and (iii) the functional annotation of target genes to metabolic pathways and/or gene ontologies using, for example, DAVID (Huang et al., Reference Huang, Sherman, Zheng, Yang, Imamichi, Stephens and Lempicki2009), KOBAS (Xie et al., Reference Xie, Mao, Huang, Ding, Wu, Dong, Kong, Gao, Li and Wei2011) programs, or the in-house tools provided by annotation databases, for example, the Gene Ontology database enrichment analysis tool (geneontology.org; Eilbeck et al., Reference Eilbeck, Lewis, Mungall, Yandell, Stein, Durbin and Ashburner2005) or the Reactome database analysis tool (reactome.org).
Function of long non-coding RNA
As it can be seen from the number of lncRNA genes or transcripts listed in Tables 1 to 3, there are more lncRNAs than mRNA molecules transcribed from a DNA template. Moreover, lncRNAs can be identified within various cell compartments, such as nucleus, nucleolus, cytoplasm and mitochondria. This reflects the variety of functions which they exhibit on cell metabolism. Technically, lncRNAs functions are related to (i) transcription through either chromatin modifications by interaction with enzymes or through interactions with transcriptional machinery proteins and miRNAs; (ii) post-transcriptional regulations of mRNA molecules, such as capping, alternative splicing, editing, transport, translation, degradation and stability; and (iii) epigenetic modifications manifested by the regulation of imprinting (Bhat and Jones Reference Bhat and Jones2016; Fernandes et al., Reference Fernandes, Guaman, Vasconcellos, Heinemann, Picardeau and Nascimento2019). On the organism level, lncRNAs are known to be abnormally expressed in many diseases with the most predominant influence on cancer and viral infections thereby playing a role of biomarkers. A practical example of lncRNA being a biomarker in human cancers is HOTAIR, whose overexpression results in development and metastases of several cancer types in humans (Lorenzi et al., Reference Lorenzi, Avila Cobos, Decock, Everaert, Helsmoortel, Lefever, Verboom, Volders, Speleman, Vandesompele and Mestdagh2019). In the healthy physiological state, lncRNAs play a role in organ differentiation during embryogenesis (Grote and Herrmann, Reference Grote and Herrmann2015) as well as in the process of aging (Xing et al., Reference Xing, Bai, Guo, Chen, Hua, Zhang, Ma, Ren, Wang and Wang2017). Most of the applications related to livestock investigate the functional annotation of lncRNAs, manifested by Gene Ontologies (GO) and KEGG pathways assigned to their target genes. These functions however are strongly related to the experimental design applied in each particular study and thus not of a universal nature. For example, in a recent study on pigs, Chen et al. (Reference Chen, Shi, Chen, Li, Li, Zou, Ch and Ch2019) applied the DAVID software to constructed clusters composed of GO terms and KEGG pathways characteristic for genes, which were targets of lncRNAs differentially expressed in relation to growth performance. In addition, You et al. (Reference You, Zhang, Liu, Song, Yang and Lian2019) applied the DAVID software for functional clustering of GO term and KEGG pathways related to target genes of lncRNAs differentially expressed in white leghorn chicken infected by the Marek’s disease virus and in a control, healthy group. In cattle, an example of functional annotation is the study of Gao et al. (Reference Gao, Li, Lai, Zhou, Wu, Huang, Lan, Lei, Chen and Dang2019). Using KOBAS, the authors tested a functional enrichment in GO terms and KEGG pathways of genes targeted by lncRNAs differentially expressed in two developmental stages of testis.
Table 1 Recent (since 2016) studies on long non-coding RNA (lncRNA), including long intergenic non-coding RNA (lincRNA), in Sus scrofa on a genome-wide scale
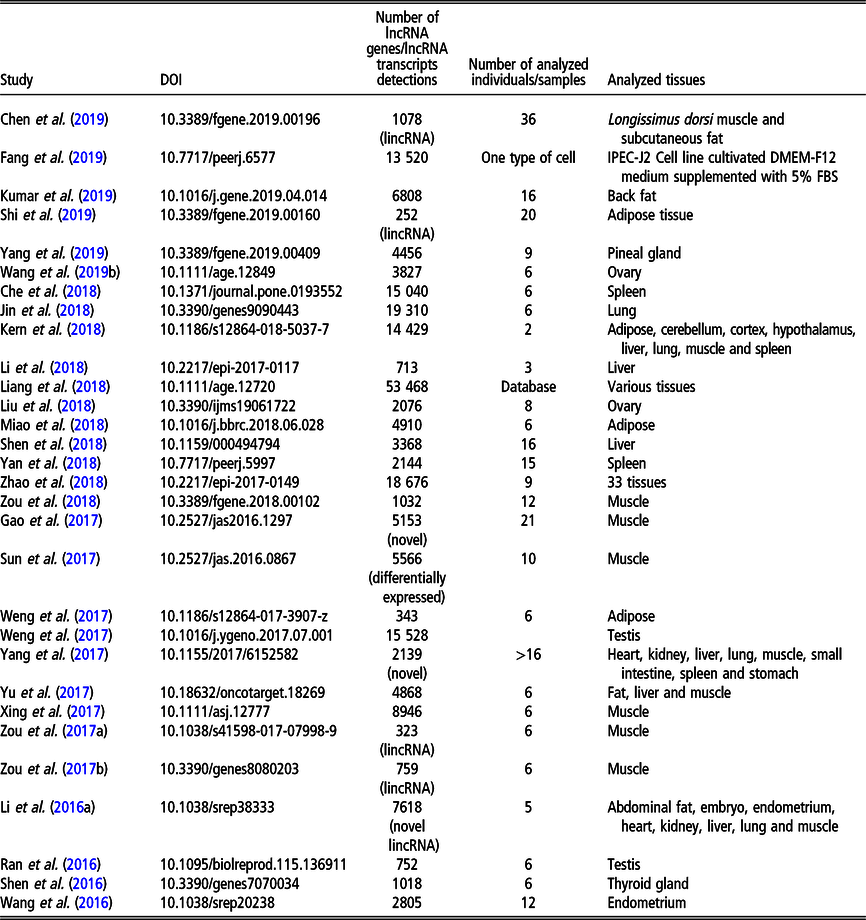
Table 2 Recent (since 2016) studies on long non-coding RNA (lncRNA), including long intergenic non-coding RNA (lincRNA), in Bos taurus on a genome-wide scale
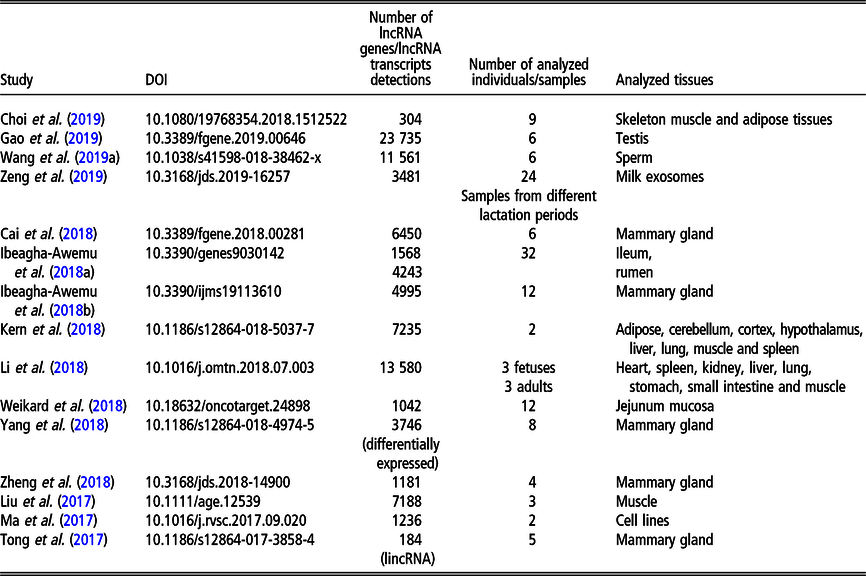
Table 3 Recent (since 2016) studies on long non-coding RNA (lncRNA), including long intergenic non-coding RNA (lincRNA), in Gallus gallus on a genome-wide scale
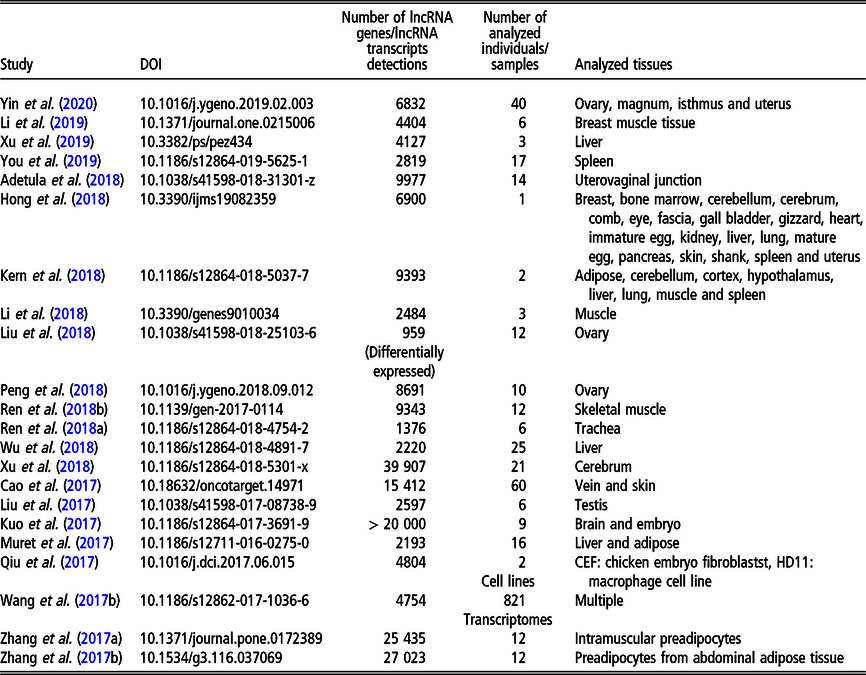
Long non-coding RNA in livestock
Sus scrofa
Among livestock, the largest number of identified lncRNA transcripts is available for pigs’ genomes, amounting to 29 585 transcripts in the NONCODE 5.0 database (Zhao et al., Reference Zhao, Li, Fang, Kang, Wu, Hao, Li, Bu, Sun, Zhang and Chen2015). Moreover, Liang et al. (Reference Liang, Yang, Li, Yu, Li, Tang and Li2018) developed the Pig LncRNANet database (lnc.rnanet.org), which stores authors’ own lncRNA discoveries, as well as results from other published studies. Although not fully mature, the database provides valuable bioinformatic functions, such as sequence BLAST, lncRNA sequence visualization including overlaps with QTL and SNV positions, as well as the visualization of transcript expression levels in various tissues. Currently, the Pig LncRNANet database contains 53 468 lncRNA records and is thereby the most comprehensive pig lncRNA catalog.
Most of the studies carried out on pigs are logically related to differential expression of all lncRNAs or only lincRNAs for traits included into the selection goal of most breeds. These comprise growth performance expressed by analyzing transcription in muscle tissue either in comparison to other tissues (Chen et al., Reference Chen, Shi, Chen, Li, Li, Zou, Ch and Ch2019), in comparison between animal groups (Zou et al., Reference Zou, Li, Luo, Li, Hu, Fu, Hou and Li2017a), between breeds with different growth performance characteristics (Gao et al., Reference Gao, Guo, Du, Cao, Yang, Pu, Wang, Zhang, Li, Jin, Wang, Liu and Li2017; Sun et al., Reference Sun, Xie, Huang, Li, Chen, Sun, Wang, Xi, Wu and Zhang2017; Yu et al., Reference Yu, Tai, Zhang, Chu, Li and Zhou2017) or between different developmental stages (Zou et al., Reference Zou, Li, Cheng, Li, Fu, Fang and Li2018). In the context of growth performance, the expression in other tissues has also been considered – intramuscular adipose tissue (Miao et al., Reference Miao, Wang, Zhang, Wei, Guo, Liu, Wang and Shi2018) or back fat tissue (Chen et al., Reference Chen, Shi, Chen, Li, Li, Zou, Ch and Ch2019; Kumar et al., Reference Kumar, Srikanth, Park, Lee, Choi, Kim, Kim, Cho, Kim, Lee, Jung, Go, Lee, Kim, Lee, Lim and Park2019). Besides growth performance, meat quality is also an important meat production trait, for which the expression of lincRNA has recently been considered, for example, by Zou et al. (Reference Zou, Li, Deng, Guan, Chen, Yuan, Xia, He, Shan and Ch2017b). Some studies analyzed the impact of testosterone deficiency on the expression of lncRNAs, either by comparing intact and castrated males (Xing et al., Reference Xing, Bai, Guo, Chen, Hua, Zhang, Ma, Ren, Wang and Wang2017; Wang et al., Reference Wang, Hua, Chen, Zhang, Bai, Gao, Li, Shi, Sheng, Gao and Xing2017a) or different male developmental stages (Ran et al., Reference Ran, Chen, Li, Wu, Liu, He, Zhang and Li2016; Sun et al., Reference Sun, Xie, Huang, Li, Chen, Sun, Wang, Xi, Wu and Zhang2017). Apart from production, lncRNA expression was also assessed for traits related to female reproduction by the comparison of expression in different developmental stages of ovaries (Liu et al., Reference Liu, Xiao, Gilbert, Cui, Zhao, Wang, Yin, Li, Zhang and Zhu2018; Wang et al., Reference Wang, Yang, Li, Li and Tang2019b) or in endometrium of pregnant and non-pregnant females (Wang et al., Reference Wang, Xue, Liu, Liu, Hu, Qiu, Zhang and Lei2016). Other experimental designs were devoted to the analysis of expression in specific organs, such as spleen (Chen et al., Reference Chen, Shi, Chen, Li, Li, Zou, Ch and Ch2019; Yan et al., Reference Yan, Huang, Sun, Yang, Shi, Jiang, Li, Wang and Gun2018) in the context of resistance against pathogenic infections, pineal (Yang et al., Reference Yang, Zhou, Li, Liu, Zhang, Ao, Li and Li2019), liver (Li et al., Reference Li, Zhang, Zhou, Wang, Liu and Liu2018) or lung (Jin et al., Reference Jin, Hu, Tu, Huang, Tang, Ma, Wang, Li, Zhou, Shuai and Li2018). Profiles of lncRNA expression in disease were considered for porcine circovirus-associated disease by Fang et al. (Reference Fang, Yang, Wang, Wang, He, Wang, Jiang and Deng2019) and for intrauterine growth restriction by Shen et al. (Reference Shen, Zhang, Li, Fu, Tang, Jiang, Li, Wang, Li, Che and Zhu2018). Moreover, instead of inter-group expression comparisons, some studies focused on identification and genomic annotation of lncRNAs in various tissues (Li et al., Reference Li, Gao, Wang, Liu, Zhang and Liu2016a; Liu et al., Reference Liu, Sun, Li, Bai, Xue, Xu, Xu, Shi, Yang and Chen2017; Yang et al., Reference Yang, Zhou, Zhu, Li, Li, Yu and Li2017; Zhao et al., Reference Zhao, Zheng, Feng, Wang, Kang, Ning, Du, Yu, Li, Zhao and Liu2018).
The most recent studies on lncRNA detection in Sus scrofa conducted on a genome-wide scale are summarised in Table 1.
Bos taurus
Although less lncRNA transcripts have been identified for cattle than for pigs, the former is the livestock species with the largest number of lncRNA genes (22 227 in NONCODE 5.0). The first genome-wide catalog of bovine intergenic lncRNAs was provided by the study of Huang et al. (Reference Huang, Long and Khatib2012) who identified 449 lncRNAs located in 405 intergenic regions, using public bovine-specific expressed sequence tag sequences. After that, the majority of studies of lncRNAs were related to their expression in the mammary gland (e.g., Cai et al., Reference Cai, Li, Liu, Zhou, Yin, Song, Zhang and Zhang2018; Yang et al., Reference Yang, Jiao, Ge, Zhang, Wang, Zhao and Wang2018; Ibeagha-Awemu et al., Reference Ibeagha-Awemu, Li, Dudemaine, Do and Bissonnette2018b) or milk exosomes (Zeng et al., Reference Zeng, Chen, Xie, Luo, He, Xi, Sun and Zhang2019) in relation to dairy production. In addition, the role of lncRNAs in beef production was addressed by assessing the expression in longissimus thoraci by Billerey et al. (Reference Billerey, Boussaha, Esquerré, Rebours, Djari, Meersseman, Kloop, Gautheret and Rocha2014), as well as in muscles and adipose tissues by Choi et al. (Reference Choi, Shin, Lee and Oh2019). Long non-coding transcriptome of male reproduction traits was analyzed by Wang et al. (Reference Wang, Yang, Guo, Zhang, Ju, Jiang, Zhao, Liu, Zhao, Wang, Sun, Wang, Zhu and Huang2019a) in sperm samples with divergent motility as well as by Gao et al. (Reference Gao, Li, Lai, Zhou, Wu, Huang, Lan, Lei, Chen and Dang2019) in different testis development stages. Among other phenotypes, Weikard et al. (Reference Weikard, Hadlich and Kuehn2013) focused on lncRNA expression in pigmented and non-pigmented bovine skin samples, Weikard et al. (Reference Weikard, Hadlich, Hammon, Frieten, Gerbert, Koch, Dusel and Kuehn2018) – on lncRNA expression dependent on energy metabolism associated with different diets and Ibeagha-Awemu et al. (Reference Ibeagha-Awemu, Do, Dudemaine, Fomenky and Bissonnette2018a) – on lncRNA expression in ileum and rumen during different developmental stages. A comprehensive genome-wide annotation of lncRNA expressed in 18 tissues was presented by Koufariotis et al. (Reference Koufariotis, Chen, Chamberlain, Vander Jagt and Hayes2015).
The most recent studies on lncRNA detection in Bos taurus conducted on a genome-wide scope are summarized in Table 2.
Gallus gallus
With 12 850 lncRNA transcripts, corresponding to 9527 genes, poultry is represented by less than half of the number of records in the NONCODE database than the above-mentioned mammalian livestock species. However, Kou et al. (Reference Kuo, Tseng, Eory, Paton, Archibald and Burt2017) comparing the complexity of human and chicken transcriptomes suggested that chicken transcriptome is similar in complexity to the human transcriptome.
During the last few years, the chicken genome has been intensively investigated in the context of lncRNA. Since meat performance is one of the economically most important polutry phenotypes, a large number of studies relate to the expression of lncRNA in tissues related to growth: muscle (Li et al., Reference Li, Ouyang, Zheng, Cai, Han, Abdalla, Nie and Zhang2016b; Cai et al., Reference Cai, Li, Ma, Wang, Han, Abdalla, Nie and Zhang2017; Ren et al., Reference Ren, Zhou, Zhou, Tian, Gu, Zhao, Chen, Han, Liu and Li2017; Li et al., Reference Li, Zhang, Zhou, Wang, Liu and Liu2018; Ren et al., Reference Ren, Fan, Liu, Wang and Zhao2018a), adipose tissue (Muret et al., Reference Muret, Klopp, Wucher, Esquerré, Legeai, Lecerf, Desert, Boutin, Jehl, Acloque, Giuffra, Djebali, Foissac, Derrien and Lagarrigue2017; Zhang et al., Reference Zhang, Zhang, Han, Zhang, Wang, Xie, Xue and Fan2017a and Reference Zhang, Zhang, Han, Zhang, Wang, Xie and Xue2017b) as well as to meat quality (e.g., Li et al., Reference Li, Li, Jiang, Zhang, Han, Jiang, Li, Tian, Yan, Kang and Sun2019). The influence of lncRNA on egg laying performance was analyzed by Peng et al. (Reference Peng, Chang, Wang, Wang, Hu, Zhao, Geng, Liu, Gong, Li, Li and Zhang2018). Another economically important one in poultry group of traits is immune response. In the context of lncRNA expression, it was analyzed by Qiu et al. (Reference Qiu, Li, Chang, Bi, Liu, Xu, Zhang, Zhao, Xu and Chen2017) and Hu et al. (Reference Hu, Chen, Jia, Xue, Dou, Dai, Xu, Sun, Geng and Cui2018) for the resistance to the Avian leukosis virus J, by Ren et al. (Reference Ren, Li, Zhou, Liu, Han, Wang, Yan, Sun, Li and Kang2018b) for the resistance to Cryptosporidium baileyi. Long non-coding RNA expression changes in the presence of selenium deficiency were addressed by Fan et al. (Reference Fan, Cao, Zhao, Shi, Zhao and Xu2017) and Cao et al. (Reference Cao, Fan, Zhao, Zhao, Yang, Zhang and Xu2017). Fertility was studied in the context of female reproduction by Liu et al. (Reference Liu, Xiao, Gilbert, Cui, Zhao, Wang, Yin, Li, Zhang and Zhu2018) and Yin et al. (Reference Yin, Lian, Zhu, Zhang, Hincke, Yang and Hou2020) for ovary, by Adetula et al. (Reference Adetula, Gu, Nwafor, Du, Zhao and Li2018) for uterovaginal tissue as well as by Yin et al. (Reference Yin, Lian, Zhu, Zhang, Hincke, Yang and Hou2020) for oviduct. Male reproduction traits were assessed by lncRNA expression study in sperm with differential motility (Liu et al., Reference Liu, Sun, Li, Bai, Xue, Xu, Xu, Shi, Yang and Chen2017). In addition, lncRNA expression study exists, which focused on the development of specific organs, such as liver (Muret et al., Reference Muret, Klopp, Wucher, Esquerré, Legeai, Lecerf, Desert, Boutin, Jehl, Acloque, Giuffra, Djebali, Foissac, Derrien and Lagarrigue2017; Wu et al., Reference Wu, Liu, Guo, Cheng, Ren, Chen, Li, Duan, Sun and Yang2018; Xu et al., Reference Xu, Zhang, Yang, Shen, Feng, Ren and Xiao2019), brain (Xu et al. Reference Xu, Che, Li, Tian, Zhu, Mishra, Dai, Li and Li2018) and ovary (Liu et al., Reference Liu, Xiao, Gilbert, Cui, Zhao, Wang, Yin, Li, Zhang and Zhu2018). Other recently analyzed phenotypes comprise lncRNA expression landscape related to chicken domestication addressed by Wang et al. (Reference Wang, Xu, Wang, Otecko, Ye, Wu and Zhang2017b) as well as the expression related to feathers and skin black color studied by Hong et al. (Reference Hong, Chai, Nam, Lim, Lee, Do, Cho and Nam2018).
The most recent studies on lncRNA detection in Gallus gallus conducted on a genome-wide scope are summarized in Table 3.
Genomic annotation of long non-coding RNAs
One of the most complete and therefore most widely used databases that store lncRNAs is the NONCODE. It provides the collection and annotation of ncRNAs, especially lncRNAs, in 17 species, including livestock. The current version of the NONCODE (5.0) contains 548 640 transcripts, identified either by the RNA-seq, expression microarrays, or based on the literature. Yet, the database is far from complete while most of the studies listed in Tables 1 to 3 report a very large number of novel lncRNAs. For instance, Kern et al. (Reference Kern, Wang, Chitwood, Korf, Delany, Cheng, Medrano, Van Eenennaam, Ernst, Ross and Zhou2018) reported that only 18.3% of pig, 1.7% of cattle and 5.7% of poultry lncRNA transcripts from the NONCODE database overlapped with transcripts detected in their analysis.
We annotated lncRNAs of five species from the NONCODE database (accessed on 20 November 2019) using the Variant Effect Predictor software (McLaren et al., Reference McLaren, Pritchard, Rios, Chen, Flicek and Cunningham2010). 51 453 lncRNA genes were annotated to the Sscrofa11.1 (Warr et al., Reference Warr, Affara, Aken, Bickhart, Beiki, Billis, Chow, Eory, Heather, Flicek, Girón, Griffin, Hall, Hannum, Hourlier, Howe, Hume, Izuogu, Kim, Koren, Liu, Manchanda, Martin, Nonneman, O’Connor, Phillippy, Gary, Rosen, Rund, Sargent, Schook, Schroeder, Schwartz, Skinner, Talbot, Tseng, Tuggle, Watson, Smith and Archibald2019; accessed from www.ensembl.org/Sus_scrofa on 20 November 2019 with the corresponding Ensembl ID GCA_000003025.6) reference genome, 25 683 lncRNA genes were annotated to the ARS-UCD1.2 reference genome of Bos taurus (Shamimuzzaman et al., Reference Shamimuzzaman, Le, Unni, Diesh, Triant, Walsh, Tayal, Connat, Hagen and Elsik2020; accessed from www.ensembl.org/Bos_taurus on 20 November 2019 with the corresponding Ensembl ID GCA_002263795.2) and 22 843 lncRNA genes were annotated to the GRCg6a reference genome of Gallus gallus (accessed from www.ensembl.org/Gallus_gallus on 20 November 2019 with the corresponding Ensembl ID GCA_000002315.5). These livestock annotations were compared to the well-annotated mouse GRCm38 (318 287 lncRNA genes) and human GRCh38 (616 532 lncRNA genes) genomes. Since genomic coordinates of lncRNAs were defined in relation to different versions of reference genomes, we converted them to the most current assembly of each species using the liftOver software (Kent et al., Reference Kent, Sugnet, Furey, Roskin, Pringle, Zahler and Haussler2002). Species-specific annotations are visualized in Figure 1. The distribution pattern of lncRNA for Homo sapiens and Mus musculus is very similar. The observed difference between those two species and the livestock species demonstrates the incompleteness of the annotation of lncRNA in livestock. Precisely, a more complete annotation of the lncRNA in humans and mice is manifested by the fact that the categories representing the actual proper annotation for lncRNA (non-coding transcript exon variant, non-coding transcript variant and regulatory region variant) make up 39.35% of all annotations in Homo sapiens and 43.84% in Mus musculus, but only 13.90% in Sus scrofa, 12.54% in Gallus gallus and as little as 3.14% in Bos taurus. In livestock, the missing ncRNA annotation seems to be predominantly assigned to intergenic and intron sequences.

Figure 1 Genomic annotation of long non-coding RNA genes from the NONCODE database for (a) human (Homo sapiens), (b) mouse (Mus musculus), (c) cattle (Bos taurus), (d) poultry (Gallus gallus) and (e) pig (Sus scrofa). UTR = Untranslated Transcribed Region.
Conclusions
The above-mentioned studies demonstrate that lncRNAs play important roles not only in the regulation of gene expression (as it was originally emphasized, see e.g., the earlier review of Mercer et al., Reference Mercer, Dinger and Mattick2009), but also in numerous other aspects of normal physiology and diseases. Compared to the number of transcripts for model organisms (Homo sapiens and Mus musculus), livestock has a relatively small number of deposited transcripts in biological databases; therefore, the main course of future research is to further improve the annotation of the non-coding part of livestock genomes. The major challenge associated with lncRNA analysis is a poor accuracy of transcript detection, which involves many data filtration stages. Since the identification workflows of lncRNAs detection change dynamically, there is a great need for defining standardized pipelines. The problem is demonstrated by a typically very large number of ‘novel’ transcripts reported by each study. As already mentioned above, Kern et al. (Reference Kern, Wang, Chitwood, Korf, Delany, Cheng, Medrano, Van Eenennaam, Ernst, Ross and Zhou2018) reported a very low overlap between the NONCODE database and transcripts detected in their analysis. Koufariotis et al. (Reference Koufariotis, Chen, Chamberlain, Vander Jagt and Hayes2015) validated 87.47% of lncRNAs expressed in liver based on samples from the same individual, but validation across animals was much lower – 55.27% of validated lncRNAs in blood. Of course, the problem with low detection accuracy is typical for all high-throughput technologies, but in the case of lncRNA detection, it is enhanced by the fact that the expression levels of lncRNAs are typically low. We need to bear in mind that such low repeatability of lncRNA detection is not necessarily always associated with false-positive or false-negative detections. The expression of lncRNA genes is not only low but also extremely tissue-specific (see e.g., graphical summary offered by the Pig LncRNANet database), related to physiological state of the individual. Moreover, since 2018, studies demonstrating a regulation of lncRNA expression by gut microbiota have emerged (Dempsey et al., Reference Dempsey, Zhang and Cui2018; Li and Cui, Reference Li and Cui2018), adding a new insight into the complexity of the transcriptome landscape.
Today’s trend is to receive economic efficiency, by fully exploiting the genetic information. That is why it is important to gain knowledge on all mechanisms controlling gene expression, such as lncRNA molecules.
Acknowledgements
None.
B. Kosinska-Selbi 0000-0003-4709-5556
M. Mielczarek 0000-0002-1086-9119
J. Szyda 0000-0001-9688-0193
Conflict of interest
None.
Ethics statement
Not applicable.
Software and data repository resources
None of the data was deposited in an official repository.




