


Implications
Male and female fertility are lowly heritable in cattle, but the reduction in generation interval achieved through the deployment of genomic selection has allowed a rapid improvement of female fertility in US dairy cattle. For several logistical reasons, genetic evaluations are not widely produced for male fertility in either beef or dairy cattle. Because the genetic correlation between male and female fertility is low, little improvement in male fertility is expected as a correlated response to selection for female fertility. To maximize food production from cattle, new approaches must be developed to improve male fertility independently of female fertility.
Introduction
In cattle, fertility is commonly measured using a number of metrics including age at puberty, calving interval, non-return rate, number of services per conception and daughter pregnancy rate (Berry et al., Reference Berry, Wall and Pryce2014). Perhaps the most useful metric for evaluating fertility in both sexes is the probability of achieving a pregnancy as a result of a single mating to a randomly sampled, but fertile, member of the opposite sex from 4 to 16 h following the onset of oestrus in the female. Because the majority of beef cattle are multi-sire mated and beef females are exposed to bulls for a period representing two to three oestrous cycles, this latter metric cannot be widely used in the beef industry. However, in the dairy industry, the majority of females are bred by artificial insemination (AI) allowing the outcomes of individual matings to be recorded. Despite this, genetic evaluations for fertility in the US dairy industry are calculated only for females. The reason for this is that when service sire is included in the genetic evaluation model, the estimated additive genetic variance for male fertility is 0 (Cole J.B, USDA ARS, personal communication) and this appears to be due to the bull studs’ use of service sire conception rate (SCR) data to titrate the number of progressively motile pre-freeze spermatozoa until a uniform non-return rate is achieved, systematically eliminating naturally occurring variation in male fertility (Van Tassell C.P., USDA ARS, personal communication; DeJarnette et al., Reference DeJarnette, Nebel and Marshall2010). As a consequence, selection occurs only for female fertility in both the US beef and dairy industries.
Although the heritabilities of male and female fertility are both generally low (Fortes et al., Reference Fortes, DeAtley, Lehnert, Burns, Reverter, Hawken, Boe-Hansen, Moore and Thomas2013a; Berry et al., Reference Berry, Wall and Pryce2014), response to selection is also governed by the extent of phenotypic variation and the length of the generation interval. The advent of genomic selection (Meuwissen et al., Reference Meuwissen, Hayes and Goddard2001) has dramatically reduced generation interval in the US dairy industry (García-Ruiz et al., Reference García-Ruiz, Cole, VanRaden, Wiggans, Ruiz-López and Van Tassell2016) and is beginning to similarly impact the US beef industry. This has enabled a remarkable increase in the rate of genetic improvement of production traits and also for female fertility within the US Holstein population, which is shown in Figure 1 (García-Ruiz et al., Reference García-Ruiz, Cole, VanRaden, Wiggans, Ruiz-López and Van Tassell2016). While there do not appear to be a large number of studies in cattle, estimates of the genetic correlation between male and female fertility is positive but generally modest in most vertebrate species (from −0.30 to 0.20 for male and female non-return rate in Danish dairy cattle, Hansen, Reference Hansen1979; ‘slight’ for male and female non-return rate in Norwegian dairy cattle, Syrstad, Reference Syrstad1981; −0.25, −0.28 and −0.41 between scrotal circumference and days to calving in Australian Hereford, Angus and Zebu crosses, respectively, Meyer et al., Reference Meyer, Hammond, Mackinnon and Parnell1991; 0.14 in Manech Tête Rousse sheep, David et al., Reference David, Bodin, Lagriffoul, Leymarie, Manfredi and Robert-Granié2007; 0.15 for male and female contributions to egg fertility in broiler chickens, Wolc et al., Reference Wolc, White, Olori and Hill2009; 0.34 for male and female contributions to conception in rabbits, Piles and Tusell, Reference Piles and Tusell2012). Consequently, the majority of genes that create variation in male fertility have male-specific functions and selection to improve female fertility will result in a positive, but less than optimal, increase in male fertility. Improvement of the overall efficiency of beef and dairy production will require the ability to identify and eliminate young bulls with sperm abnormalities and unacceptable semen quality (Taylor et al., Reference Taylor, Schnabel and Sutovsky2018) and the development of predictors of genetic merit for male fertility that may be applied within and perhaps also across breeds. In this manuscript, we address the current state of knowledge concerning genetic variants responsible for variation in male fertility and the approaches that should be taken to enable the improvement of male fertility in beef and dairy cattle.
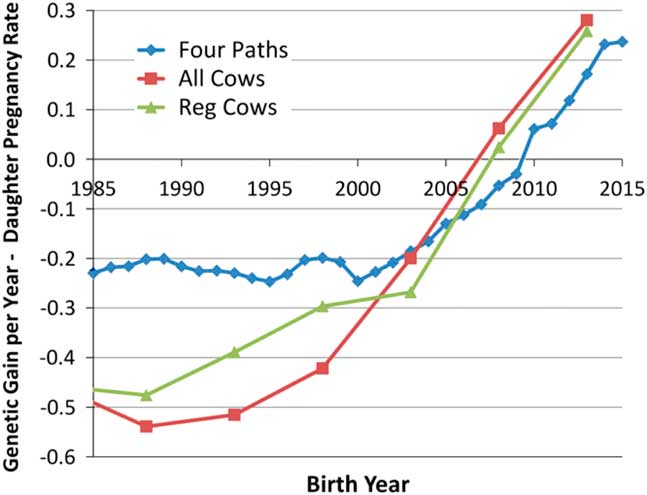
Figure 1 (colour online) Average genetic gain per year for female fertility in US Holsteins (García-Ruiz et al., Reference García-Ruiz, Cole, VanRaden, Wiggans, Ruiz-López and Van Tassell2016).
Mendelian variants causing variation in male and female fertility
Of the loci that create genetic variation in both male and female fertility, the most obvious are loss-of-function (LOF) mutations in genes that are essential for life. In the human genome, 7168 (33.3%) of the 21 556 annotated genes are essential for life (Chen et al., Reference Chen, Lu, Chen, Zhao and Bork2017) meaning that the functionality of at least one copy of each of these genes is required for human life. The number and proportion of essential genes in cattle are probably very similar to those in humans. Mutations which disrupt the functionality of the proteins encoded by essential genes are LOF mutations and for genes located on the autosomes (non-sex chromosomes), homozygosity for a LOF mutation, or heterozygosity of two chromosomes each with a different LOF mutation in the same gene leads to lethality. Because these mutations are transmitted to progeny by both males and females, they are responsible for variation in genetic merit for both male and female fertility. The majority of LOF mutations produce early embryonic loss due to failure to implant or develop. These pregnancy losses are frequently not noticed, but calf losses may also occur in the second and third trimesters of pregnancy or postnatally, manifesting as a genetic defect. These loci are subject to purifying selection because homozygotes are removed every generation leading to relatively small decreases in allele frequency in each successive generation. However, the frequency of some of these LOF lethal alleles can be driven to high levels in a population by the extensive use of AI, which allows carrier bulls to transmit what might otherwise be a rare LOF mutation to a large number of progeny.
Because there is a large number of genes that are essential and these are all targets for mutation, there may be a very large number of lethal LOF mutations within a population. However, the frequency of the majority of these alleles is generally very low and the joint effect of these rare alleles on the mean fertility of the population is small. Because relatively few bulls have been whole genome sequenced (less than 4000 world-wide), the majority of rare variants are yet to be found, either because they were not present in the sequenced animals or because they were detected only once in a sequenced animal and filtered as potentially being a sequence error. Of those that have been discovered, not all can confidently be predicted to be a LOF mutation. A mutation that produces a charged amino acid substitution that is predicted to not be tolerated may, or may not, result in the LOF of a protein. Several lethal LOF mutations have been found in cattle by applying the haplotypic insufficiency analytical technique first described by VanRaden et al. (Reference VanRaden, Olson, Null and Hutchison2011). Using this technique, high-density single nucleotide polymorphism (SNP) genotypes such as those produced by the 54 001 SNP BovineSNP50 assay (Matukumalli et al., Reference Matukumalli, Lawley, Schnabel, Taylor, Allan, Heaton, O’Connell, Moore, Smith, Sonstegard and Van Tassell2009) are first phased so that for each genotyped individual, the two alleles present at each SNP genotype are assigned in a specific order to each of the chromosomes that are present in that individual. Each specific combination of SNP alleles present on a chromosome or chromosomal segment is called a haplotype and the specific pair of haplotypes present within each individual is called a diplotype. Next, the frequencies of haplotypes and diplotypes present in a sample of genotyped animals are tallied for small chromosome segments of, say, 20 consecutive SNPs. The probability of observing no individuals that are homozygous for each haplotype is calculated based on the sample size and the assumption of random inheritance of haplotypes from each parent (Hardy–Weinberg equilibrium). To identify genomic regions that are likely to harbour an autosomal recessive lethal LOF allele, the method of VanRaden et al. (Reference VanRaden, Olson, Null and Hutchison2011) identifies haplotypes that never occur in homozygous form when we would expect to see homozygotes in the sample based on the frequency of the haplotype. The logic behind this method is that if all of the chromosomes in the population that are identified by the same haplotype of 20 SNP alleles harbour a LOF lethal mutation, then every homozygote must be lethal and these individuals will never be seen in the population. Figure 2 provides a schematic representation of this process that shows that when 100 000 individuals have been genotyped and the chromosomes have been phased, 20 marker haplotypes that are at a frequency of 2% in the genotyped sample would be expected to be observed in homozygous form in 40 individuals. When in practice none are observed, the probability that this is due to chance alone is vanishingly small (P HWE=[1−p 2] N =4×10−18) and we may conclude that the reason that we did not observe any homozygotes is that the haplotype harbours an autosomal recessive lethal LOF allele. Clearly the approach is limited by the need for many genotyped individuals, otherwise low frequency haplotypes will not appear in homozygous form simply due to chance. The approach also assumes that all chromosomes with the same haplotype identified based on alleles at 20 SNPs contain the lethal allele. This may not be the case when the mutation has recently occurred, in which case chromosomes in some individuals will carry the lethal mutation while chromosomes in other individuals will not carry the lethal allele despite the fact that the haplotypes all appear to be identical based upon the marker information. In this case, we will observe fewer homozygotes than expected based on the haplotype frequency, but certainly more than none. This reduces the power of the test and therefore much larger sample sizes are necessary to detect homozygote deficiency when the lethal allele is not perfectly associated with a single haplotype.

Figure 2 (colour online) Two haplotypes (coloured yellow or blue) defined by alleles present at 20 consecutive single nucleotide polymorphism markers (locations shown as horizontal bars) are present at frequencies of P=0.98 and q=0.02 in a genotyped sample of size n=100 000 cattle. On average, 40 animals would be expected to be homozygous for the rare (blue) haplotype.
A slightly different version of this test was discussed by VanRaden et al. (Reference VanRaden, Olson, Null and Hutchison2011) and implemented by Hoff et al. (Reference Hoff, Decker, Schnabel and Taylor2017) that capitalizes on the available pedigree information in genotyped cattle populations. When trios of sire, dam and progeny or patrios of sire, maternal grandsire and progeny have all been genotyped and a particular haplotype is never observed in homozygous form, we can count the number of trios in which the sire and dam are both heterozygotes or the number of patrios where the sire and maternal grandsire are both heterozygotes and calculate the probability of not observing a progeny that is homozygous for the rare haplotype. This is demonstrated in Figure 3 for the case of a patrio where the dam has not been genotyped, the frequency of the rare haplotype in the population is q and the son has a probability of (2q+1)/8 of being homozygous aa for the rare haplotype ‘a’ but is observed to be AA or Aa. If N such patrios in which the sire and maternal grandsire are both heterozygotes are counted as never producing a homozygous progeny, the probability of this being due to chance alone is P P =[0.75–0.25q] N and for haplotypes at a frequency of 2% in the population P P is 1.6×10−13 for as few as n=100 patrios. This may represent many fewer than 300 individuals as sires and maternal grandsires may be common across many patrios. The probability calculation requires only independent assortment of parental alleles in each family and not independent families. The approach analyses either overlapping or non-overlapping 20 SNP marker windows sequentially along each chromosome in order to scan the entire genome for the presence of lethal mutations and so there is a multiple testing problem that requires adjustment of the probability values produced for each test to appropriately manage the false discovery rate.

Figure 3 (colour online) An example of a cattle patrio in which the sire and maternal grandsire are both heterozygous for a rare haplotype ‘a’ at a frequency q=1−p in the sample and for which no homozygotes are observed. The probability that the son is homozygous for the a haplotype when it does not carry a lethal mutation is (2q+1)/8.
Cole et al. (Reference Cole, VanRaden, Null, Hutchison, Cooper and Hubbard2017) reported 26 recessive haplotypes that are currently tracked in the US dairy breed genomic evaluation system. Of these, two are fertility related in each of Jersey, Brown Swiss and Ayrshire and eight are fertility related in Holsteins (Table 1). The frequencies of these 12 lethal haplotypes range from 0.37% to 13% and average 11.4%, 7.2%, 1.97% and 6.7% in Ayrshire, Brown Swiss, Holstein and Jersey, respectively. The causal mutations responsible for embryonic lethality have been discovered for only nine of the 14 haplotypes. Fritz et al. (Reference Fritz, Capitan, Djari, Rodriguez, Barbat, Baur, Grohs, Weiss, Boussaha, Esquerre, Klopp, Rocha and Boichard2013) performed a genome-wide scan in European dairy breeds for homozygous haplotype deficiency using 47 878 Holstein, 16 833 Montbeliarde and 11 466 Normande animals genotyped with the BovineSNP50 and found 18 haplotypes in Holstein, 11 in Montbeliarde and six in Normande with frequencies ranging from 1.7% to 9%. Nine of these haplotypes were found to be associated with reductions in fertility when directly tested against conception rate in both heifers and adult cows using heterozygous trio matings, validating the presence of lethal alleles. An additional eight haplotypes were associated with conception rate in heifers or adult cows. Whole genome sequence data from 25 Holstein, 11 Montbeliarde and nine Normande bulls with important individual contributions to their respective breeds (from 1.1% to 10.8%), sequenced to a depth of coverage from 8.9 to 39.2× were investigated in an attempt to identify the deleterious mutations associated with eight of the haplotypes associated with fertility in heifers and cows, leading to strong candidates for two lethal mutations in SHBG and SLC37A2 in the Montbeliarde. Six of the recessive lethal haplotypes detected in French Holsteins coincide with those segregating in the US Holstein population (Cole et al., Reference Cole, VanRaden, Null, Hutchison, Cooper and Hubbard2017) which is expected considering the broad international use of US bulls. Sahana et al. (Reference Sahana, Nielsen, Aamand, Lund and Guldbrandtsen2013) identified 17 homozygote deficient haplotypes at frequencies of from 1.4% to 3.4% in 7937 Nordic Holsteins genotyped with the BovineSNP50 BeadChip in an analysis in which haplotypes were based on 25 consecutive SNPs. These haplotypes appeared to define eight genomic regions likely to harbour lethal alleles, and of these, six regions were confirmed as having effects on fertility when tested for associations with either non-return rate or calving interval. Remarkably, of all the candidate lethal mutations found in Nordic Holsteins, only the locus on chromosome 21 fully overlaps the locus responsible for Brachyspina in US Holsteins (Cole et al., Reference Cole, VanRaden, Null, Hutchison, Cooper and Hubbard2017; Table 1).
Table 1 Haplotypes or mutations responsible for embryonic lethality discovered by genome-scanning for haplotype or genotype homozygote insufficiency

1 Online Mendelian inheritance in animals. Taxon ID 9913 represent cattle.
2 Multiple listed genes represent a deletion.
3 Bos taurus chromosome.
4 Cole et al. (Reference Cole, VanRaden, Null, Hutchison, Cooper and Hubbard2017).
5 Fritz et al. (Reference Fritz, Capitan, Djari, Rodriguez, Barbat, Baur, Grohs, Weiss, Boussaha, Esquerre, Klopp, Rocha and Boichard2013).
6 Sahana et al. (Reference Sahana, Nielsen, Aamand, Lund and Guldbrandtsen2013).
7 Charlier et al. (Reference Charlier, Li, Harland, Littlejohn, Coppieters, Creagh, Davis, Druet, Faux, Guillaume, Karim, Keehan, Kadri, Tamma, Spelman and Georges2016).
8 Kadri et al. (Reference Kadri, Sahana, Charlier, Iso-Touru, Guldbrandtsen, Karim, Nielsen, Panitz, Aamand, Schulman, Georges, Vilkki, Lund and Druet2014).
9 Sahana et al. (Reference Sahana, Iso-Touru, Wu, Nielsen, de Koning, Lund, Vilkki and Guldbrandtsen2016).
10 Hoff et al. (Reference Hoff, Decker, Schnabel and Taylor2017). Haplotypes not validated as fertility associated.
Pausch et al. (Reference Pausch, Schwarzenbacher, Burgstaller, Flisikowski, Wurmser, Jansen, Jung, Schnieke, Wittek and Fries2015) performed a genome-wide scan for homozygous haplotype deficiency in 25 544 Fleckvieh cattle using the Illumina BovineSNP50 BeadChip and found four haplotypes (identified as FH1 through FH4) that were deficient in their observed numbers of homozygotes. Two haplotypes were never observed in homozygous form and the frequencies of all four haplotypes ranged from 2.9% to 4.1%. Insemination success was reduced by 6.64% and 5.99% in FH1 and FH4 carrier-to-carrier matings, respectively. A 4.06% decline in insemination success and a 4.3% reduced first-year survival rate of progeny was observed for FH3 carrier-to-carrier matings. Insemination success and stillbirth rate were not affected in FH2 carrier-to-carrier matings; however, juvenile mortality in progeny was increased by 6.6% compared with the survival of progeny from non-risk matings. Using whole genome sequence data from 263 animals from ten different cattle breeds (including 145 Fleckvieh and 15 Simmental) with an average of 10× sequence coverage, these authors identified strong functional candidate mutations underlying two of the haplotypes. A small indel producing a frameshift in SLC2A2 was shown to activate cryptic splice sites in the processed mRNA leading to aberrant splicing at exon 7, while a missense mutation in SUGT1 was predicted to be highly damaging to SGT1 protein function. With many fewer genotyped animals, Hoff et al. (Reference Hoff, Decker, Schnabel and Taylor2017) analysed BovineSNP50 data for 3961 registered Angus animals and identified seven haplotypes genome-wide that were predicted to harbour autosomal recessive lethal alleles. These were not validated to directly affect fertility or survival rates but ranged in frequency from 2.3% to 7.6%. Despite an analysis of sequence data from 109 bulls resequenced to an average 27× depth of coverage from which 1 to 27 bulls (average of 11.4 bulls were sequenced for each haplotype) were predicted to be carriers of each recessive lethal haplotype, no strong candidates for any of the lethal mutations were found despite six of the regions being detected as harbouring from 1 to 118 concordant (never homozygous) mutations. Because Hoff et al. (Reference Hoff, Decker, Schnabel and Taylor2017) restricted their attention to SNPs, many of the causal variants may be small or large insertions or deletions that are not easily or reliably detected from variant calling pipelines.
Advantages of the haplotype-based analysis approach are that it does not require any reproductive data and can be accomplished as a by-product of genotyping animals to enable genomic selection. As very large numbers of animals are genotyped within each breed (over 2 million Holsteins https://www.cdcb.us/Genotype/cur_density.html and over 300 000 registered Angus https://www.angus.org/pub/newsroom/releases/062717-single-step.html animals have now been genotyped in the United States) it becomes possible to detect haplotypes containing autosomal recessive lethal mutations that are at very low frequency within a population. For example, Holstein Haplotype 4 (HH4) is at a frequency of only 0.37% in the US Holstein population (Cole et al., Reference Cole, VanRaden, Null, Hutchison, Cooper and Hubbard2017). The disadvantage of the approach is that haplotype-based selection is possible only when all chromosomes possessing the haplotype signature harbour the lethal allele leading to the complete absence of homozygotes for the haplotype. For recent mutations, where the lethal allele exists only on some of the chromosomes possessing the haplotype that has a homozygous deficiency, additional family analyses must be performed to identify which families transmit the lethal version of the haplotype and which transmit the viable haplotype. The problem is that the analysis does not actually identify which mutation within the genomic region spanned by the haplotype is the cause of the lethal phenotype. To accomplish this requires sequencing individuals that carry the lethal haplotype to identify candidate mutations that can then be directly tested by genotyping within the population for the absence of homozygotes. Fritz et al. (Reference Fritz, Capitan, Djari, Rodriguez, Barbat, Baur, Grohs, Weiss, Boussaha, Esquerre, Klopp, Rocha and Boichard2013) warn that the existence of strong linkage disequilibrium may lead to the causative variants actually being located outside of the intervals defined by the haplotypes. This appears to be supported by the results in Table 1 which summarize studies performed in Holstein cattle world-wide and reveal that lethal haplotypes detected in different Holstein strains may not completely overlap, although we presume that the same causal variant was detected.
Mesbah-Uddin et al. (Reference Mesbah-Uddin, Guldbrandtsen, Iso-Touru, Vilkki, De Koning, Boichard, Lund and Sahana2018) utilized 10× average depth of coverage whole genome sequence data produced for 67 Holsteins, 27 Jerseys and 81 Nordic Red Cattle to identify 8480 large deletions (199 bp to 773 kb; mean 4.5 kb; median 1 kb). The deletions were validated to have an overall false discovery rate of 8.8% using Illumina BovineHD genotype intensity data produced for 26 of the sequenced Holsteins, chromosome breakpoint assembly and alignment and the sequencing of PCR amplicons spanning breakpoints. By examining the deletion genotypes of the sequenced individuals, the authors found 5000 deletions for which at least one sequenced individual was homozygous. Among these were 167 deleted genes that were demonstrated to be non-essential based on the occurrence of live homozygote individuals. This study is essentially the reciprocal of the haplotype-based approaches just discussed as it can unequivocally identify genes to be non-essential via the existence of living animals that are homozygous knockouts. While the resolution at which candidate lethal mutations are scanned is dramatically increased by considering whole genome sequence variation, the identification of putative lethal mutations is limited by the relatively small sample of sequenced individuals. However, a ~525 kb deletion on chromosome 23, which is known to cause stillbirth in Nordic Red Cattle (Sahana et al., Reference Sahana, Iso-Touru, Wu, Nielsen, de Koning, Lund, Vilkki and Guldbrandtsen2016) was among the haplotypes that were not found in homozygous form in the sequenced animals.
Kadri et al. (Reference Kadri, Sahana, Charlier, Iso-Touru, Guldbrandtsen, Karim, Nielsen, Panitz, Aamand, Schulman, Georges, Vilkki, Lund and Druet2014) performed a genome-wide association study (GWAS) in 10 099 bulls from five European dairy breeds using estimated breeding values of the bulls for an index of cow fertility traits and found 14 quantitative trait loci (QTLs) at genome-wide significance, of which the most significant was located on chromosome 12. By repeating the analysis within each of the breeds, they identified that this QTL was primarily segregating in Finnish Ayrshire and Swedish Red, but was not detectable in Holstein-Friesian, Danish Red or Jerseys. A haplotype-based analysis identified a single haplotype (A27) that was found to contain a 660 kb deletion that contained four genes, that produced an increase in milk production in heterozygous animals but that was an embryonic lethal in homozygotes likely due to the loss of RNASEH2B. The frequency of this deletion haplotype was 6.5%, 11.5% and 16% in Danish, Swedish and Finnish Red Cattle, respectively.
Charlier et al. (Reference Charlier, Li, Harland, Littlejohn, Coppieters, Creagh, Davis, Druet, Faux, Guillaume, Karim, Keehan, Kadri, Tamma, Spelman and Georges2016) resequenced the genomes of 496 Holstein-Friesian×Jersey animals and 50 Belgian Blue Cattle to an average depth of 11× and resequenced the exomes of 78 animals from six Bos taurus breeds to an average depth of 40×. A total of 186 112 exonic variants, including 1377 stop-gain, 112 stop-loss, 3139 frameshift, 1341 splice-site, 85 338 missense, and 92 163 synonymous variants were discovered. Of the missense variants, 22 939 were predicted by SIFT and/or PolyPhen to be disruptive or damaging to protein function. From these, 3779 candidate variants (frameshift, splice-site, stop-gain and missense variants predicted to be damaging and/or deleterious) for embryonic loss were genotyped in ~35 000 New Zealand dairy cattle (296 LOF and 3483 missense) and 1050 were genotyped in ~6300 Belgian Blue cattle (108 LOF, 942 missense). From the produced genotype data, the authors estimated that 15.5% of the tested LOF variants and 5.9% of the tested missense variants were embryonic lethal mutations. Nine common LOF variants were confirmed to be embryonic lethal mutations based upon the absence of homozygotes in carrier×carrier matings (Table 1).
Table 1 contains a list of haplotypes or mutations recently discovered via genome scans for homozygote insufficiency that have been demonstrated to affect fertility in several beef and dairy breeds. There are several remarkable observations from the data in this table. First, the average frequency of lethal mutations detected in cattle populations to date is about 3.8%, which is much larger than you would expect for loci subjected to strong purifying selection. This is clearly due to the extensive use of AI in these populations, which results in the strong enrichment of alleles present within the genomes of selected bulls that go on to have large numbers of progeny. However, the number of lethal loci detected to date in numerically large breeds such as Holstein (17) is far greater than for the numerically smaller breeds such as Jersey (3), Brown Swiss (2) and Ayrshire (2). Furthermore, the average frequency of lethal alleles in Holsteins (2.4%) is considerably lower than in Jersey (6.66%), Brown Swiss (7.23%) and Ayrshire (11.4%). This likely reflects a greater number of genotyped animals used to detect a larger number of rarer alleles, but it also may suggest that the larger number of bulls used in AI in numerically large breeds allows a greater enrichment of lethal loci in the breed, albeit at lower individual frequencies. While it is difficult to make conclusions about the identities of mutations based on haplotype data (Charlier et al. (Reference Charlier, Li, Harland, Littlejohn, Coppieters, Creagh, Davis, Druet, Faux, Guillaume, Karim, Keehan, Kadri, Tamma, Spelman and Georges2016) found three lethal mutations located within a 1.82 Mb region of chromosome 11 in two breeds) the data in Table 1 suggest that the majority of the common lethal alleles are breed specific. That is, these mutations occurred after breed formation and were driven to relatively high frequencies within the respective breeds by the use of selective breeding. On the other hand, there is also evidence for potentially different lethal loci segregating within subpopulations of Holsteins. Despite the widespread use of high merit US Holstein bulls world-wide, the loci found by Sahana et al. (Reference Sahana, Nielsen, Aamand, Lund and Guldbrandtsen2013) in Nordic Holsteins are not completely consistent with those of Fritz et al. (Reference Fritz, Capitan, Djari, Rodriguez, Barbat, Baur, Grohs, Weiss, Boussaha, Esquerre, Klopp, Rocha and Boichard2013) for French Holsteins which are largely consistent with those in the summary of Cole et al. (Reference Cole, VanRaden, Null, Hutchison, Cooper and Hubbard2017) for US Holsteins. This would be expected if the majority of lethal mutations are quite recent in origin and if there is subdivision, possibly caused by different selection objectives in each of the subpopulations.
Cole et al. (Reference Cole, Null and VanRaden2016) estimated that the economic losses due to reduced fertility and perinatal calf death in US Holsteins was almost US$ 11 million per year and Charlier et al. (Reference Charlier, Li, Harland, Littlejohn, Coppieters, Creagh, Davis, Druet, Faux, Guillaume, Karim, Keehan, Kadri, Tamma, Spelman and Georges2016) estimated the cost due to embryonic losses from nine confirmed lethal loci to be NZ$ 13.8 million in New Zealand dairy cattle and €2.7 million in Belgian Blue cattle. The use of available genotype data to detect and avoid carrier×carrier matings is clearly an effective way to improve the fertility of cattle, but may become difficult to implement as the number of detected lethal mutations and genetic defects increases within individual breeds. Cole (Reference Cole2015) has suggested an alternative approach in which the estimated genetic merits of individuals for net merit are adjusted for the economic losses due to fetal losses and has shown the method to be effective at reducing the frequency of recessive lethal alleles, whilst maintaining current rates of genetic improvement. Lethal alleles are also excellent candidates for multiplex genome editing in which the heterozygous deleterious alleles are simply removed from the genomes of the bulls that are placed into AI each generation (Hickey et al., Reference Hickey, Bruce, Whitelaw and Gorjanc2016).
Non-Mendelian effects on male fertility
Loss-of-function lethal mutations behave in a manner that is called Mendelian inheritance because the ability of an allele to impact the viability of a progeny is the same if the allele is transmitted through the male or female germ lines. However, loci that are transmitted through the mitochondrial genome (or genomes in cases of heteroplasmy; about 5% of individuals within mammalian species appear to have at least two mitochondrial genomes), on the Y chromosome, or on the X chromosome can cause patterns of inheritance that are non-Mendelian. There are also cases of variants that are located on the non-sex associated autosomes that lead to phenotypes that are inherited in a non-Mendelian fashion. Imprinted loci are transmitted through the germ-line with either alleles transmitted by females being silenced (female imprinting in which case the male inherited allele is expressed) usually by DNA methylation or alleles transmitted by males being silenced (male imprinting). Each generation, the methylation status of these alleles is reset according to the sex of individual. Flisikowski et al. (Reference Flisikowski, Venhoranta, Nowacka-Woszuk, McKay, Flyckt, Taponen, Schnabel, Schwarzenbacher, Szczerbal, Lohi, Fries, Taylor, Switonski and Andersson2010) reported the segregation of a lethal locus in which the mode of inheritance appeared to be autosomal dominant with incomplete penetrance (not all individuals possessing the lethal allele die). Semen from a Finnish Ayrshire bull was used to artificially inseminate 1900 Finnish Ayrshire heifers and cows and field data collected by the AI Cooperative suggested that 42.6% of the late pregnancies attributed to the bull ended in stillbirths or abortions. This figure is close enough to a 50 : 50 segregation ratio to suggest that the bull was heterozygous for a mutation that was transmitted as a dominant lethal. The fact that lethality was slightly <50% suggested that there could have been some progeny that inherited the lethal allele that did not die, presumably because they were protected by variants at other loci and this might explain why the bull itself escaped death. However, the causal variant was shown to be a 110 kb deletion within the MIMT1 gene, which is part of the PEG3 (paternally expressed gene) domain. As the alleles transmitted by males are expressed (the maternal alleles are silenced by methylation), both copies of MIMT1 were transmitted to progeny in activated form but progeny that inherited the deletion allele had no functional MIMT1 expression as all MIMT1 copies transmitted by their dams were silenced. The fact that the bull survived the inheritance of a lethal mutation was because he inherited the deletion MIMT1 allele in a silenced form from his dam and the functional MIMT1 allele was inherited from his sire. Thus, LOF mutations in MIMT1 have absolutely no effect on female fertility but are lethal when transmitted by the sire. MIMT1 encodes a non-coding RNA, which is not translated into a protein and has an unknown function. However, this naturally occurring mutation demonstrates that MIMT1 is essential to life. Magee et al. (Reference Magee, Sikora, Berkowicz, Berry, Howard, Mullen, Evans, Spillane and MacHugh2010) have also shown that SNPs within the PEG3 gene cluster are associated with calving, calf performance and fertility traits in Irish Holstein-Friesian cattle.
The bovine Y chromosome comprises a small pseudo-autosomal region with a homolog on the X chromosome and a much larger male-specific region which contains clusters of genes thought to be essential for male reproduction because they are primarily expressed during testicular development (Yang et al., Reference Yang, Chang, Yasue, Bharti, Retzel and Liu2011; Chang et al., Reference Chang, Yang, Retzel and Liu2013). The number of genes on the bovine Y chromosome appears to be surprisingly large relative to the number of genes found on the human Y chromosome suggesting a much greater potential role of the Y chromosome in phenotype determination, particularly male fertility, in cattle than in humans (Chang et al., Reference Chang, Yang, Retzel and Liu2013). However, there appears to have been surprisingly little work conducted to date to characterize the effects of mutations in bovine Y chromosome genes on male fertility. This is probably due to the fact that until recently, there was no reference sequence assembly for the bovine Y chromosome and due to the highly repetitive and palindromic nature of the Y chromosome sequence, the existing assembly is of much lower quality than the assembly for the autosomal genome. One study has shown that the number of copies of members of the HSFY and ZNF280BY gene families varies by almost an order of magnitude in cattle (from ~20 to over 300 copies) and that variation in copy number of both families was negatively correlated with testis size, but positively correlated with SCR (Yue et al., Reference Yue, Dechow, Chang, DeJarnette, Marshall, Lei and Liu2014). Because the number of copies of members of both families within individual bulls is positively correlated (Yue et al., Reference Yue, Dechow, Chang, DeJarnette, Marshall, Lei and Liu2014), it is not clear which family (if either) was causal for effects on male fertility. Copy number variation in members of the PRAMEY gene family encoding proteins found in the sperm head and tail has been shown to be negatively correlated with percentage of normal sperm and non-return rate, but not with SCR in Holstein bulls (Yue et al., Reference Yue, Chang, DeJarnette, Marshall, Lei and Liu2013).
Mutations within genes or regulatory regions on the X chromosome have the potential to be severely deleterious to fertility in males who have only a single copy of the X chromosome and far less so in females who have two copies of the X chromosome, although one copy is presumably randomly inactivated in each cell within every tissue. Because of this, and like Y chromosome mutations, these variants are exposed to extremely strong purifying selection in males. Consequently, we might expect to find many fewer X and Y chromosome mutations affecting fertility than autosomal mutations. Despite this, using a GWAS in indicine and indicine×taurine bulls, Fortes et al. (Reference Fortes, Reverter, Kelly, McCulloch and Lehnert2013b) found that the majority of genome-wide associations for scrotal circumference and percentage of normal spermatozoa at 24 months of age were located on the X chromosome. De Camargo et al. (Reference De Camargo, Porto-Neto, Kelly, Bunch, McWilliam, Tonhati, Lehnert, Fortes and Moore2015) examined the effects of seven SNPs responsible for amino acid substitutions in seven genes located in regions of the X chromosome previously identified by the GWAS of Fortes et al. (Reference Fortes, Reverter, Kelly, McCulloch and Lehnert2013b) in the same experimental population and detected significant associations for SNPs in LOC100138021, CENPI and TAF7L with percentage of normal spermatozoa (Table 2) and for SNPs in TEX11 and AR with scrotal circumference. None of the SNPs detected as being associated with male fertility were found to be associated with female fertility. A brief overview of effects of chromosomal aberrations and structural variation on male fertility is presented in Supplementary Material S1.
Table 2 Genomic regions identified by genome-wide association analysis or candidate gene analysis as associated with sire conception rate

SNP=single nucleotide polymorphism.
1 SNP located within or near identified gene or window contains the listed genes.
2 Lan et al. (Reference Lan, Peñagaricano, DeJung, Weigel and Khatib2013).
3 Estimated relative conception rate.
4 Khatib et al. (Reference Khatib, Monson, Huang, Khatib, Schutzkus, Khateeb and Parrish2010).
5 PCR amplicon contains identified SNP but exact reference genome position not provided.
6 Han and Peñagaricano (Reference Han and Peñagaricano2016).
7 QTL validated in two or more studies.
8 rs number corresponds to marker name but provided chromosomal coordinates are not correct.
9 Peñagaricano et al. (Reference Peñagaricano, Weigel and Khatib2012).
10 Validated in follow-up analysis of expanded genotyped sample.
Quantitative trait loci responsible for variation in male fertility
The evolution of DNA methylation probably occurred as a method to silence the transcriptional activity of retrotransposons that were integrated into the germ-line via retroviruses (Nagamori et al., Reference Nagamori, Kobayashi, Shiromoto, Nishimura, Kuramochi-Miyagawa, Kono and Nakano2015). DNA methylation appears to be ubiquitous in mammalian genomes; however, variation in the extent of DNA methylation at specific loci is known to occur and this variation has been associated with the quantitative regulation of gene expression. By examining the methylation profiles of DNA extracted from the spermatozoa of bulls with high- and low-conception rates, Verma et al. (Reference Verma, Rajput, De, Kumar, Chakravarty and Datta2014) found differentially methylated regions associated with 151 genes with functions in germ cell development, spermatogenesis, capacitation, and embryonic development in water buffalo. Using a similar approach in Holstein bulls identified as being extreme for SCR based upon at least 300 inseminations, Kropp et al. (Reference Kropp, Carrillo, Namous, Daniels, Salih, Song and Khatib2017) found 76 genomic regions to be differentially methylated in the DNA extracted from the spermatozoa of high and low-SCR bulls. What is not clear from these epigenetic studies is whether the detected differential methylation is the cause of differences in fertility or the effect of some other genomic mechanism that is responsible for the aberrant methylation of sequences regulating the expression of genes that are required for high fertility. Similarly, several studies have found differences in the spermatozoa mRNA transcript (Card et al., Reference Card, Krieger, Kaproth and Sartini2017), miRNA and piRNA (Capra et al., Reference Capra, Turri, Lazzari, Cremonesi, Gliozzi, Fojadelli, Stella and Pizzi2017) and protein (Peddinti et al., Reference Peddinti, Nanduri, Kaya, Feugang, Burgess and Memili2008) abundances. While these approaches have been pursued from the perspective of developing biomarkers of male fertility, the capability of the differentially abundant molecules to predict variation in male fertility has yet to be established.
There is an intrinsic relationship between GWAS studies and genomic selection. In GWAS, a large number of markers approximately evenly spread throughout the genome are assayed in a sample of phenotyped individuals and maker effects are individually tested to identify those that meet a pre-specified statistical threshold. This approach identifies the largest effect associations within the genome, based on the available sample size, and ignores the potential myriad of markers with small effects on the trait. These large effect markers can be used to generate molecular estimates of genetic merit, but typically these explain only small percentages of the overall additive genetic variance. In other words, there are relatively few markers of large effect and a much larger (but unknown) number of small effect markers. On the other hand, genomic selection attempts to use all of the markers (or a reasonably large subset of the markers when some Bayesian analyses are utilized) to predict genetic merit, capturing many more of the small effect variants, and can produce estimates of genetic merit that frequently explain at least 70% of the additive genetic variance in traits. In terms of application, it has historically been simpler and less expensive to genotype 10 or 20 markers in a large sample of individuals than many thousands of markers. However, with the recent deployment and very rapid adoption of high-density SNP arrays for genotyping in cattle and the impact that genomic selection has had on the improvement of female fertility in US Holsteins (García-Ruiz et al., Reference García-Ruiz, Cole, VanRaden, Wiggans, Ruiz-López and Van Tassell2016), it is clear that the development of predictions of genetic merit for SCR should be a priority in cattle genomics. Two limitations to the approach are that all of the early industry genotyping was performed using the Illumina BovineSNP50 assay which did not include Y chromosome variants (although all of the newer assays do), and the currently utilized statistical analyses do not appropriately model the effects of X chromosome markers (Taylor, Reference Taylor2014). Imprinted loci are probably approximately correctly modelled in analyses of data from only a single sex, but are not correctly modelled in analyses of data from both sexes.
Feugang et al. (Reference Feugang, Kaya, Page, Chen, Mehta, Hirani, Nazareth, Topper, Gibbs and Memili2009) estimated conception rates for 874 US Holstein bulls with an average of 788 breedings (range 101 to 11 997) in a probit analysis after adjusting for herd-year-month, parity, cow, days in milk and sire proven status. The 10 highest and 10 lowest fertility bulls (mean difference 15.4% in SCR) were genotyped using the Affymetrix/ParAllele 9919 SNP GeneChip and 8207 polymorphic markers were analysed in a single marker regression of allele dosage on each bull’s SCR phenotype. The four most strongly associated SNPs (P<0.0001; Table 2) were then genotyped in a larger cohort of 100 low- and 101 high-fertility bulls and the SNPs on chromosomes 1 and 4 were validated as being associated with SCR (P<0.05). Peñagaricano et al. (Reference Peñagaricano, Weigel and Khatib2012) performed a GWAS in 1755 Holstein bulls with SCR data using 38 650 SNPs with minor allele frequencies >5% using a linear model correcting for relatedness among bulls and testing the effects of SNPs individually either fit with genotypes as additive, or additive and dominance effects. After correcting for multiple testing, they found eight SNPs defining five separate QTLs associated with SCR. Han and Peñagaricano (Reference Han and Peñagaricano2016) performed a GWAS using 44 449 estimated SCR animal effects (additive genetic+non-additive genetic and permanent environment effects; VanRaden P., USDA ARS, personal communication) available on 10 884 US Holstein bulls. Of these animals, 7447 had high-density SNP genotype data and after filtering markers with minor allele frequencies <1% or that were sex-linked, 58 029 autosomal markers were analysed. A single-step BLUP analysis was used incorporating pedigree information for the animals that had not been genotyped. The percentage of additive genetic variance in SCR explained by all of the SNPs within 1.5 Mb genomic regions was estimated and significant regions were declared when they explained at least 0.5% of the SCR additive genetic variance. The authors also performed a single-SNP analysis in which the mixed model included the effects described above but only for the genotyped animals and found results that were consistent with those from the single-step BLUP. Han and Peñagaricano (Reference Han and Peñagaricano2016) estimated that the SNPs explained 32% of the variance in SCR animal effects and, consequently, 68% of the unexplained variance was due to additive genetic variance not captured by the SNPs, and the service sire non-additive genetic and permanent environmental effects. Han and Peñagaricano (Reference Han and Peñagaricano2016) also found six SCR QTLs located on chromosomes 5, 13, 21 and 25 that explained at least 0.5% of the SCR additive genetic variance (Table 2).
As all three studies were conducted in US Holstein bulls, we should expect considerable consistency among the results and also for candidate gene studies previously reporting significant associations with SCR (Khatib et al., Reference Khatib, Monson, Huang, Khatib, Schutzkus, Khateeb and Parrish2010; Lan et al., Reference Lan, Peñagaricano, DeJung, Weigel and Khatib2013). However, Table 2 shows that only two loci were consistently detected in at least two different studies in US Holsteins. Associations with RIMS1 on chromosome 9 between 11.8 and 12.1 Mb were detected by Feugang et al. (Reference Feugang, Kaya, Page, Chen, Mehta, Hirani, Nazareth, Topper, Gibbs and Memili2009) and Han and Peñagaricano (Reference Han and Peñagaricano2016). Associations were also detected on chromosome 25 in the region from 0.9 to 4.7 Mb by Peñagaricano et al. (Reference Peñagaricano, Weigel and Khatib2012) and Han and Peñagaricano (Reference Han and Peñagaricano2016). This is somewhat typical of GWAS studies particularly when they are underpowered and there are a few variants of large effect responsible for trait variation. Druet et al. (Reference Druet, Fritz, Sellem, Basso, Gérard, Salas-Cortes, Humblot, Druart and Eggen2009) performed a GWAS with 148 microsatellite markers in 10 families containing 515 French Holstein bulls for semen production phenotypes of ejaculated volume and sperm concentration, number of spermatozoa, motility, velocity, percentage of motile spermatozoa after thawing and abnormal spermatozoa. Of the 11 detected QTL, only two affecting ejaculated volume (chromosome 15 at 22 cM) and sperm motility (chromosome 7 at 34 cM) appear to overlap with the SCR QTL reported in Table 2.
Abdollahi-Arpanahi et al. (Reference Abdollahi-Arpanahi, Morota and Peñagaricano2017) used the SCR data for 7447 bulls that was employed by Han and Peñagaricano (Reference Han and Peñagaricano2016) for GWAS to evaluate the utility of genomic selection to predict the genetic merit of AI bulls for SCR using a fivefold cross-validation scheme and SNP feature selection. Using all 54 706 fitted SNP, the average correlation between predicted genetic merit and SCR phenotype was 0.34 corresponding to a prediction accuracy of ~0.63. Selecting SNPs within genes with Gene Ontology and Medical Subject Heading terms including reproduction, fertilization, sperm motility or sperm capacitation did not improve prediction accuracy. However, restricting the analysis to the 18 659 SNPs detected as being associated (P nominal<0.05) with SCR increased the correlation slightly to 0.35. Nonlinear models uniformly outperformed linear models for accuracy of prediction, but the improvement was generally fairly small.
Conclusions
Male and female fertility are positively correlated but the correlation is low and genetic predictions for fertility are currently only produced for females. Genomic selection has produced dramatic increases in female fertility in a relatively short period of time in US Holsteins demonstrating that a low heritability is not the sole determinant of selection response. While this improvement should also have produced a small correlated response in male fertility, this is an unsatisfactory solution considering the economic importance of fertility to cattle production and the need to increase the efficiency and quantity of animal-based food proteins world-wide.
In dairy cattle, there is an opportunity to rapidly develop genomic predictions for male fertility (in both sexes) considering the large number of genotyped animals and the availability of SCR phenotypes. However, these phenotypes should be based on inseminations made by yearling bulls in which sperm dosages have been standardized and this will require collaboration between AI organizations. In the US beef industry, the majority of genetic improvement in all traits is created by selection within the registered sector. Despite the reduced use of AI relative to the dairy industry, it should similarly be possible to capture the benefits of increased rates of genotyping to develop genetic predictions for SCR. The increasing use of sexed semen to produce heifer replacements within the US dairy industry also presents an opportunity for the generation of SCR data for beef bulls, as sexed male semen from beef bulls is increasingly being used to breed dairy cows that were not selected to produce heifer replacements. Scoring conception rates in dairy cows is agnostic to the breed of bulls used in AI. Finally, increasing the rate of use of AI in commercial beef herds via the use of synchronization of oestrus and ovulation to facilitate fixed-time AI of beef cows has an enormous opportunity for the collection of SCR phenotypes in beef bulls. If 10% of the commercial beef cows in the US were bred by AI, the industry could collect more SCR phenotypes than are currently produced within the entire dairy industry. If genomic predictions of merit for male fertility are to be produced for both males and females, efforts should be invested to develop and evaluate models that appropriately model the effects of sex chromosome and imprinted variants.
Acknowledgements
The authors appreciate the support of NIH-USDA Dual Purpose with Dual Benefit Program grant number NIH 1R01HD084353. PS is also supported by USDA-NIFA grant 2015-67015-23231 and by seed funding from the Food for the 21st Century Program of the University of Missouri. J.T. and R.S. are supported by USDA-NIFA grants 2013-68004-20364, 2016-67015-24923 and 2017-67015-26760. The authors also appreciate the constructive criticisms of two anonymous reviewers who appreciably improved the manuscript.
Declaration of interest
The authors have no conflicts of interest.
Ethics statement
No animals were used in this review study.
Software and data repository resources
No software or unpublished data were used in this review.
Supplementary Material
To view supplementary material for this article, please visit https://doi.org/10.1017/S1751731118000599







