


Introduction
Beef consumers increasingly seek meat of high and consistent quality. As a consequence, the beef industry is investing in research aimed at the identification of indicators of quality and an increased understanding of muscle biology to control quality traits.
Beef quality includes sensory quality traits (tenderness, flavour, juiciness, colour, etc), nutritional value, healthiness and technological quality (Geay et al., Reference Geay, Bauchart, Hocquette and Culioli2001) and also other issues such as animal welfare, environmental concerns, traceability, etc (Hocquette and Gigli, Reference Hocquette, Gigli, Hocquette and Gigli2005). Whereas the latter are more complex or subjective issues, the former are directly associated with muscle biology traits of live animals and during post-mortem processing treatments.
Biotechnology is widely regarded as one of the most promising life science frontiers for the next decade. Together with informatics, biotechnology is expected to lead to revolutionary changes in our society and economy. This scientific revolution, sometimes referred to as the genomic revolution, is global and is creating new opportunities in all biological sciences including medicine, human health and nutrition, agronomy and, the subject of this paper, animal science.
This paper aims to illustrate how animal scientists can take advantage of the recent developments in functional genomics to get a better understanding of the factors which determine gene expression and hence muscle physiological traits controlling beef eating quality.
‘Genomics’: definition, history, potential benefits and limitations
Definitions and history of genomics
In the past, molecular biologists studied one gene at a time in isolation from the larger context of other genes. This was particularly true in physiology where only a few genes characterising a specific metabolic pathway were studied together. In more recent times, high-throughput analyses of gene expression have allowed a global view of gene expression thanks to the development of techniques such as DNA arrays and proteomic approaches (Hocquette, Reference Hocquette2005).
The first high-throughput molecular biology methods to be developed were sequencing methods. Therefore, sequencing of genomes from many species has been achieved or is in progress. This has facilitated a better understanding of the genome structure and the identification of polymorphisms (mainly single nucleotide polymorphisms or SNP), which allow more precise gene mapping. In beef science, this will contribute to the more rapid identification of genetic markers which will be useful for animal breeding, with the objective to improve beef quality traits as previously reviewed (Kühn et al., Reference Kühn, Leveziel, Renand, Goldammer, Schwerin, Williams, Hocquette and Gigli2005).
Now, the application of genomics to biology is gradually evolving from the discovery of genetic determinants (structural genomics) to ‘biological signatures’ (functional genomics). The latter is a typical pattern of gene expression, or even a typical profile of proteins or metabolites characterising a specific physiological or pathological state. The methods to determine these biological signatures are currently reshaping biology and are referred to as transcriptomics, proteomics and metabolomics/metabonomics (Hocquette, Reference Hocquette2005). Functional genomics therefore describes a new scientific field mid way between genetics and physiology. The current challenge for biological research is to integrate structural and functional genomics (Tuggle et al., Reference Tuggle, Dekkers and Reecy2006) and also to associate data from the different ‘-omic’ sciences with phenotypic data (Hocquette, Reference Hocquette2005). This process is facilitated by rapid developments in computational biology or bioinformatics, which has followed the rapid expansion in genomic research. Several analytical approaches, bioinformatic methods and tools have indeed been suggested to integrate data, for instance expression data at mRNA and protein levels (Cox et al., Reference Cox, Kislinger and Emili2005) in association with metabolite profiling (Weckwerth et al., Reference Weckwerth, Wenzel and Fiehn2004) to identify real biochemical networks. More generally, bioinformatics is becoming crucial for the analysis of expression data with the ultimate objective to extract biologically meaningful information from the lists of differentially expressed genes. A variety of bioinformatic tools are available for data-mining depending on the question being asked (Hanai et al., Reference Hanai, Hamada and Okamoto2006). Many experts indicated that bioinformatics and computational biology approaches will be major areas of emphasis in the next few years in both research and practical usage even for livestock (Henderson et al., Reference Henderson, Thomas and Da2005).
Potential benefits of genomics
The development of the new genomic high-throughput technologies is reshaping biology due to four new paradigms in science. The first one of these paradigms, can be described as a shift away from characterising a small number of key molecules (such as high differentially expressed genes or proteins), but on molecular signatures (cluster of genes, of proteins, of metabolites, etc), which are characteristic of a biological process or of a specific phenotype. This shift has been made possible with the advent of high-throughput methods of data acquisition and of bioinformatics methods to handle large amounts of data.
Another new paradigm is the change in research conception. Up to now, hypothesis-driven research has been conducted with the aim of testing new biological hypotheses which have emerged from previous studies or theoretical considerations. High-throughput genomic techniques are capable of uncovering associations between previously unknown molecules (DNA, RNA, proteins, metabolites) or previously uncharacterised DNA/protein sequences and physiological traits of interest. Genomics is thus capable of generating new biological hypotheses which can then be further studied by more focused approaches. This will impact most markedly on the characterisation of complex traits, which are governed by interactions between many genes with small effects. An example of such a complex trait is beef tenderness, which depends on connective tissue characteristics, lipid content, fibre composition and meat ageing, all complex biological phenomena in themselves.
The third new paradigm is the goal of capturing the structure-function relationships of any genome as a whole. Gene and protein studies, as well as metabolomics, will provide the link between the genome and the biological processes which give rise to the phenotype. The most important characteristics of biological organisms are their ability to change in response to changing environments. Genomics (and especially functional genomics) will thus help to understand physiology, in addition to promoting progress in genetics (Andersson and Georges, Reference Andersson and Georges2004).
The last important new paradigm associated with genomics is that high-throughput gene interrogation systems will help to integrate knowledge about genes associated with different physiological functions. Knowing what genes are activated following stress by the environment, infections by pathogens and increasing production levels of milk or meat will enable researchers to assess interactions between all these biological mechanisms. This will help to develop novel management and breeding strategies that simultaneously promote animal health, well-being, food quality and safety.
In medicine, genomics will be used for effective prediction of a patient's disease risk and drug reactions, and the interaction between both. Preventive medicine and medical therapy will be personalised. The development of genomic applications for personalised medicine will require associations of gene marker profiles and gene expression patterns with clinical data (Hocquette, Reference Hocquette2005).
In agriculture and animal science, the outcomes of genomics will include improvements in food safety, in crop yield, in traceability and in quality of animal products (dairy products and meat) through increased efficiency in breeding and better knowledge of animal physiology. DNA-based techniques have been developed for the detection of bacterial contamination (beef safety), for species and animal identification (breed and individual traceability), and for genetic selection (genetic improvement of beef quality). On the other hand, RNA- and protein-based techniques may one day be used to characterise the health, physiological and nutritional status of the animals according to their breed, their genetic type, the rearing factors or the place where they are bred. These techniques may also be adapted to characterise each muscle type. For all these reasons, genomic techniques may contribute to quality control of beef. Protein-based techniques may be employed to monitor protein degradation during post-mortem ageing of beef, a process which controls tenderness. Therefore, genomics is likely to play a major role in sustainable agriculture due to the wide range of its potential applications (Harlizius et al., Reference Harlizius, van Wijk and Merks2004). The current knowledge indicates that the major genes causing variability for similar traits in different species or breeds are rarely the same. Therefore, the study of the species of interest is needed even if research in farm animals strongly relies on the genomic knowledge in other species such as humans and model organisms (Harlizius et al., Reference Harlizius, van Wijk and Merks2004).
Limitations of genomics
Many of the limitations to genomics research are of a technical nature. The technical limitations of expression array technology are still under active discussion (Shields, Reference Shields2006; Sievertzon et al., Reference Sievertzon, Nilsson and Lundeberg2006). Array technology is based on the principle that probes (cDNA from expressed sequence tag (EST) libraries or oligonucleotides) can be arrayed on membranes or glass slides for hybridisation with labelled targets, which originate from the biological samples of interest. The flatness of the glass support makes it possible to miniaturise the procedure. The resulting images of hybridisation are captured and analysed using software that quantifies the signal of each spot. The intensity of each spot is proportional to the amount of mRNA of the corresponding gene expressed in the studied sample.
The major technical problems that have affected expression array technology are associated with the spotted cDNA arrays which were the first forms of DNA expression arrays available and which are now almost superseded by spotted oligo arrays or manufactured oligo arrays. Problems with cDNA arrays apply generally to all species and range from the availability and inconsistent quality of molecular probes to be printed on the arrays and difficulties in array preparation (inconsistent fidelity across arrays, low specificity or low concentration of printed probes) and utilisation (Murphy, Reference Murphy2002; Drobyshev et al., Reference Drobyshev, Hrabé de Anegelis and Beckers2003). Commercial microarray platforms, particularly those that employ in situ synthesis of oligomer probes to construct arrays are now available for the bovine and are reducing these sources of variation to very low levels. Consistent protocols for array experimentation (RNA extraction from samples, labelling, and hybridisation) and data acquisition (image capture and normalisation procedures) are emerging and microarrays are considered a reliable method for quantitating gene expression data (Canales et al., Reference Canales, Luo, Willey, Austermiller, Barbacioru, Boysen, Hunkapiller, Jensen, Knight, Lee, Ma, Maqsodi, Papallo, Peters, Poulter, Ruppel, Samaha, Shi, Yang, Zhang and Goodsaid2006).
The statistical analysis of genomic data is a challenge shared by all genomics researchers. The challenge begins with the design of large animal experiments that nevertheless contain sufficient biological replication to allow the discovery of gene expression associations with a high level of confidence. The animal genomics community has contributed to the development of microarray analysis methods by adapting the use of mixed model analysis to microarray experimentation (Reverter et al., Reference Reverter, Byrne, Bruce, Wang, Dalrymple and Lehnert2003; Pfister-Genskow et al., Reference Pfister-Genskow, Myers, Childs, Lacson, Patterson, Betthauser, Goueleke, Koppang, Lange, Fisher, Watt, Forsberg, Zheng, Leno, Schultz, Liu, Chetia, Yang, Hoeschele and Eilertsen2005). In proteomics, research by INRA scientists has also allowed improvement in statistical analyses by adapting the SAM (significance analysis of microarray) method (Meunier et al., Reference Meunier, Bouley, Piec, Bernard, Picard and Hocquette2005) and clustering methodologies (Meunier et al., Reference Meunier, Dumas, Piec, Hébraud, Béchet and Hocquette2007) to two-dimensional gel electrophoresis experiments.
A last challenge is to share, within the bovine community, the genomic data related to beef quality (from genetic information, transcriptomics and proteomics to phenotypes). Consequently, it is now accepted that the comparison of results from different laboratories requires standardisation procedures which have been proposed for both transcriptomic (Brazma et al., Reference Brazma, Hingamp, Quackenbush, Sherlock, Spellman, Stoeckert, Aach, Ansorge, Ball, Causton, Gaasterland, Glenisson, Holstege, Kim, Markowitz, Matese, Parkinson, Robinson, Sarkans, Schulze-Kremer, Stewart, Taylor, Vilo and Vingron2001) and proteomic (Taylor et al., Reference Taylor, Paton, Garwood, Kirby, Stead, Yin, Deutsch, Selway, Walker, Riba-Garcia, Mohammed, Deery, Howard, Dunkley, Aebersold, Kell, Lilley, Roepstorff, Yates, Brass, Brown, Cash, Gaskell, Hubbard and Oliver2003) analyses. Methods to jointly analyse microarray experimentation have been developed for a large set of bovine muscle microarray results (Reverter et al., Reference Reverter, Barris, Moreno-Sánchez, McWilliam, Wang, Harper, Lehnert and Dalrymple2005). In addition, methods have been developed for computing networks of correlated gene expression from bovine muscle gene expression data (Reverter et al., Reference Reverter, Hudson, Wang, Tan, Barris, Byrne, McWilliam, Bottema, Kister, Greenwood, Harper, Lehnert and Dalrymple2006).
Powerful data management tools and computational techniques are also required to store, analyse, and compare the increasing amount of genomic information in cattle. The number of expressed sequenced tags in NCBI's GenBank has now surpassed 1 million for cattle. Other specific bioinformatic databases have been developed.
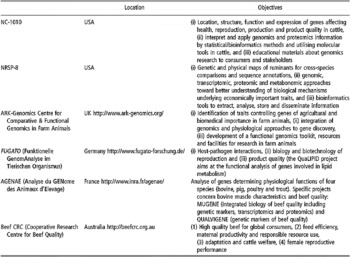
Some of the existing databases and associated bioinformatic tools are devoted specifically to applications within livestock species, especially cattle (Table 1). For instance, most of the single-locus traits in livestock are listed in the OMIA (Online Mendelian Inheritance in Animals) database. Other databases have been constructed in UK (Hu et al., Reference Hu, Mungall, Law, Papworth, Nelson, Brown, Simpson, Leckie, Burt, Hillyard and Archibald2001), Australia (Hawken et al., Reference Hawken, Barris, McWilliam and Dalrymple2004) and France (Table 1).
Table 1 Some specific bioinformatics tools and browsers for bovine genomics (extracted, adapted and updated from Fadiel et al. (Reference Fadiel, Anidi and Eichenbaum2005))

Bovine genome sequencing and applications for breeding
Bovine genome sequence
Sequencing of the bovine genome started in December 2003. It is under way at the Baylor College of Medicine Sequencing Centre in Houston (Texas, USA).
As set out below, the availability of the bovine genome sequence will be useful for both agricultural purposes and human health. Accordingly, the contributors to the $53 million international effort were: the National Human Genome Research Institute, USDA (NRI and ARS), State of Texas, Genome Canada, Kleberg Foundation, Commonwealth Scientific and Industrial Research Organisation (CSIRO) Australia, New Zealand, United States Beef Council, Texas Beef Council and South Dakota Beef Council.
The animal whose DNA was sequenced is a Hereford cow, named L1 Dominette 0144. The second draft of the bovine genome sequence has been deposited into free public databases. The current assembly (Btau 3.0, August 2006) is based on a 7.1-fold coverage of the bovine genome. The sequence data can be accessed via GenBank at NIH's National Centre for Biotechnology Information (NCBI), or via EMBL Bank at the European Molecular Biology Laboratory's Nucleotide Sequence Database and via the DNA Data Bank of Japan. A number of browsers have been developed for the bovine genome: NCBI's Map Viewer, UCSC Genome Browser at the University of California at Santa Cruz, the Ensembl Genome Browser at the Wellcome Trust Sanger Institute in Cambridge, England and the Ruminant-Human Genome Browser at CSIRO Australia (Table 1).
Sequencing at lighter coverage, to derive additional SNP, is being carried out in other breeds, such as the Holstein, Angus, Jersey, Limousin, Norwegian Red and Brahman and there are plans to also sequence the bovine Y chromosome.
In a white paper (Gibbs et al., Reference Gibbs, Weinstock, Kappes, Loren and Womack2004), the rationale and objectives of the bovine genomic sequencing initiative were set out. Eight different biological objectives were considered. The first one is to improve human health. Understanding genetic interactions with environmental factors will indeed be a major aim of medical research in the next decades. The bovine is a valuable model to achieve this goal because a vast amount of research has been and will be conducted to study the genetic × environment interaction in this species in the case of multigenic physiological traits. The second objective is informing mammalian biology. The bovine is indeed a good model for understanding endocrinology, developing reproductive techniques and studying reproductive or metabolic diseases. For example, different cattle breeds have different capacity to deposit fat. Detailed knowledge of the molecular regulation of this trait is important for both the production of marbled beef and the understanding of obesity and diabetes in human beings. The third and fourth objectives of the bovine genome sequencing are to contribute to the annotation of the human genome sequence and to provide additional links between the sequence of humans and other species by comparative genomics. This was recently done by a high-resolution whole-genome cattle-human comparative map: 3 204 ordered markers in the bovine genome over a total of 3 484 are anchored in the human genome. The comparative coverage relative to the human genome is about 91%. This will facilitate identification of candidate genes for economically important traits (Everts-van der Wind et al., Reference Everts-van der Wind, Larkin, Green, Elliott, Olmstead, Chiu, Schein, Marra, Womack and Lewin2005). The fifth objective is to expand our knowledge of the biological mechanisms related to health. It is suggested that alternative transcripts and post-translational modifications must play a major role in phenotypes. The phenotypic diversity of many cattle breeds throughout the world is a tremendous resource to study the molecular mechanisms which control tolerance to pathogens, altitude or heat tolerance, susceptibility to any type of illness, etc. The sixth and seventh objectives are to provide surrogate systems for human experimentations and to facilitate the ability to do experiments, especially in genetics. The last objective is to contribute to the understanding of evolutionary processes. To summarise, it is expected that the bovine genome sequencing, and more generally, any genomic work in cattle will have an impact not only on the livestock industries, but also on all other mammalian research, especially research into human genetics and health.
Current knowledge of genetic markers in bovine
The practical genetic issues of genomics have been described elsewhere (Renand et al., Reference Renand, Larzul, Le Bihan-Duval and Le Roy2003; Smith et al., Reference Smith, Thallman, Casas, Shackelford, Wheeler and Koohmaraie2003; Kühn et al., Reference Kühn, Leveziel, Renand, Goldammer, Schwerin, Williams, Hocquette and Gigli2005). In a few words, the objective of breeders is to exploit the variability among animals by selecting those with a superior genetic potential for the trait of interest. Many traits suitable for improvement using quantitative genetic methods are easy or inexpensive to measure on a great number of animals, but many of the important traits are not. Several of the meat quality traits, for example, are difficult to improve, such as meat tenderness and marbling, so researchers are currently looking for genes that influence genetic potential for these traits to select animals. This is a promising approach because genotyping is now increasingly easy and inexpensive to perform. One important benefit of high-throughput techniques of genomics will indeed be the identification and the practical use of polymorphisms within DNA sequences of those genes (Garnier et al., Reference Garnier, Klont and Plastow2003; Barendse, Reference Barendse2005). Single nucleotide polymorphisms (SNP) are the most frequent form of DNA variation in mammals. They are abundant, normally biallelic and easy to detect by automatic techniques.
So far, polymorphisms in some genes have been reported for their association with tenderness or beef marbling (i.e. the amount of visual intramuscular fat) (Barendse, Reference Barendse1997; Reference Barendse2001 and Reference Barendse2003 and Barendse et al., Reference Barendse, Bunch, Thomas, Armitage, Baud and Donaldson2004). They include the genes encoding μ-calpaïn (Page et al., Reference Page, Casas, Heaton, Cullen, Hyndman, Morris, Crawford, Wheeler, Koohmaraie, Keele and Smith2002; White et al., Reference White, Casas, Wheeler, Shackelford, Koohmaraie, Riley, Chase, Johnson, Keele and Smith2005) and lysyl oxidase and calpastatin (Barendse, Reference Barendse2001; Drinkwater et al., Reference Drinkwater, Li, Lenane, Davis, Shorthose, Harrison, Richardson, Ferguson, Stevenson, Renaud, Loxton, Hawken, Thomas, Newman, Hetzel and Barendse2006) for tenderness. They also include the genes encoding leptin (Buchanan et al., Reference Buchanan, Fitzsimmons, van Kessel, Thue, Winkelman-Sim and Schmutz2002), DGAT1 (Thaller et al., Reference Thaller, Kühn, Winter, Ewald, Bellmann, Wegner, Zühlke and Fries2003), TG (Barendse et al., Reference Barendse, Bunch, Thomas, Armitage, Baud and Donaldson2004), RORC (Barendse, Reference Barendse2003), GH1 (Schlee et al., Reference Schlee, Graml, Rottmann and Pirchner1994), SCD, mitochondria (Mannen et al., Reference Mannen, Kojima, Oyama, Mukai, Ishida and Tsuji1998; Reference Mannen, Morimoto, Oyama, Mukai and Tsuji2003), mitochondrial transcription factor A (Jiang et al., Reference Jiang, Kunej, Michal, Gaskins, Reeves, Busboom, Dovc and Wright2005) and FABP4 (Michal et al., Reference Michal, Zhang, Gaskins and Jiang2006) for marbling. Summaries of the tests have been published (e.g. Dekkers, Reference Dekkers2004; Jeon et al., Reference Jeon, Lee, Kim, Park and Oh2006) although no listing of markers is comprehensive. Other efforts are devoted to the identification of SNP in a large set of candidate genes which are associated with meat quality traits measured in 15 different European breeds. This is the EU-funded project (GeMQual, http://www.gemqual.org/). So far, a total of about 710 SNP have been identified in 209 genes and these SNP are being genotyped in 450 bulls from the 15 breeds. The expected results are SNP that may have an effect on the carcass, muscle or meat quality traits (Hocquette et al., Reference Hocquette, Renand, Levéziel, Picard and Cassar-Malek2006). Table 2 contains a list of all the commercially available DNA tests for cattle, which can be obtained from web searches, although there are more tests in the literature than there are tests being offered to farmers.
Table 2 Commercialised DNA tests for cattle QTL

† Multiple tests are those based on several genes, although some tests are not based on sequences directly associated with genes, and for some tests the identity of the sequence has not been revealed.
‡ Year is year commercialised.
§ First commercialiser, some of the tests, such as those based on CAPN1, CAST and LEP are now offered by several laboratories.
There are other discoveries of DNA markers but these are not yet available as commercialised DNA tests.
Tests for Casein and Lactoglobulin, originally milk protein variants, now DNA tests, not included in this Table, these were originally described and worked on in studies from the 1960s to mid 1984, see for example Genmark AG.
Table does not include (1) colour mutations, (2) disease mutations, (3) mutations for discrete traits such as poll/horn.
Many of these associations have been confirmed in independent studies, although some have failed to be confirmed. Confirmations for the calpain and calpastatin genes have been reported by several authors (Page et al., Reference Page, Casas, Quaas, Thallman, Wheeler, Shackelford, Kochmaraie, White, Bennett, Keele, Dikeman and Smith2004; Casas et al., Reference Casas, White, Wheeler, Shackelford, Koohmaraie, Riley, Chase, Johnson and Smith2006; Morris et al., Reference Morris, Cullen, Hickey, Dobbie, Veenvliet, Manley, Pitchford, Kruk, Bottema and Wilson2006; Schenkel et al., Reference Schenkel, Miller, Jiang, Mandell, Ye, Li and Wilton2006). For marbling several of the genes have been confirmed in some studies while in others some tests have failed to be confirmed (Thaller et al., Reference Thaller, Kühn, Winter, Ewald, Bellmann, Wegner, Zühlke and Fries2003; Nkrumah et al., Reference Nkrumah, Li, Basarab, Guercio, Meng, Murdoch, Hansen and Moore2004; Barendse et al., Reference Barendse, Bunch and Harrison2005; Kononoff et al., Reference Kononoff, Deobald, Stewart, Laycock and Marquess2005; Nkrumah et al., Reference Nkrumah, Li, Yu, Hansen, Keisler and Moore2005; Pollak, Reference Pollak2005; Schenkel et al., Reference Schenkel, Miller, Ye, Moore, Nkrumah, Li, Yu, Mandell, Wilton and Williams2005; Barendse et al., Reference Barendse, Bunch, Harrison and Thomas2006; Rincker et al., Reference Rincker, Pyatt, Berger and Faulkner2006). Marbling is a more difficult trait to work with than tenderness, since it is a visual inspection of the carcass and hence is a measurement with a greater error, which means that it will require larger samples than normal to be detected. The failure to confirm an association usually occurs in samples where there are fewer than 1000 animals in the study. At present there are no guidelines on the composition of confirmatory studies, and different approaches are being taken in different countries. Most of the confirmations are ad hoc, however, using whatever samples are to hand. In some confirmations, the commercialiser is asked what samples they want used to evaluate the gene. Such approaches are unsatisfactory since they can lead to the type II error of accepting a null hypothesis as true when it is false, due to inadequacies in the experimental design, primarily due to sample sizes being too small. Clearly, sample sizes need to be large enough to ensure that this sort of error is minimised (Barendse, Reference Barendse2005).
A number of high throughput SNP genotyping platforms have recently become commercially available for the bovine. The MegAllele™ Genotyping Bovine 10K SNP Panel assays approximately 10 000 SNP discovered by the Bovine Genome Sequencing Project and the CSIRO. A 32 000 SNP update of this panel is about to be commercially released. Similarly, the bovine BeadChip™ genotyping system allows around 10 000 bovine SNP to be assayed in parallel. A new version of the bovine iSelect BeadChip™ is under development in collaboration with the USDA-ARS, the University of Missouri-Columbia and the University of Alberta. This genotyping platform will allow the analysis of 12 samples in parallel on a single microarray with over 48 000 SNP per sample. Research groups all over the world are currently applying these genotyping tools to populations of cattle extensively phenotyped for beef and dairy production and health traits. In the future, more than 1 million SNP within the Bos taurus breeds and well over 2 million with the Brahman animal included are predicted by comparing the Hereford genome assembly with traces from other breeds (Womack, Reference Womack2006).
Rapid progress in discovering patterns of genetic variation that can predict animal performance can be expected from these international efforts. The increased knowledge of functional candidate genes, which will be discovered from functional genomics approaches (array data, proteomic studies) is expected to further contribute to the progress of this field.
Henderson et al. (Reference Henderson, Thomas and Da2005) predict that there are three challenges currently facing the beef industry with the use of genetic markers in genetic selection: (i) the adequate collection of DNA marker information by breed associations, (ii) quality control mechanisms for use and interpretation of DNA marker information, and (iii) increased expertise to use genetic markers discovered by the scientific communities. Furthermore, the improvement of the efficiency of marker-assisted selection does require the characterisation of regulatory sequences of gene expression, the development of methods to rapidly screen genomes of multiple individuals, a better understanding of gene functions and interactions, and more emphasis on complex traits with low heritabilities.
Available molecular tools for transcriptomic studies
Tissue-specific cDNA arrays for beef quality research
Before the development of bovine cDNA microrrarrays, some groups used available human microarrays in cross-species hybridisation studies. For instance, RNA from cattle skeletal muscle (Sudre et al., Reference Sudre, Leroux, Piétu, Cassar-Malek, Petit, Listrat, Auffray, Picard, Martin and Hocquette2003 and Reference Sudre, Cassar-Malek, Listrat, Ueda, Leroux, Jurie, Auffray, Renand, Martin and Hocquette2005a) and oocyte (Dalbies-Tran and Mermillod, Reference Dalbies-Tran and Mermillod2003) have been hybridised on human cDNA microarrays. However, because cross-species hybridisation may generate spurious results, researchers have paid careful attention to data analysis. Therefore, interesting results were obtained despite loss of information associated with the technical constraints.
Many different research teams throughout the world have published the construction of bovine cDNA libraries from various tissues including liver and intestine (Dorroch et al., Reference Dorroch, Goldammer, Brunner, Kata, Kühn, Womack and Schwerin2001; Herath et al., Reference Herath, Shiojima, Ishiwata, Katsuma, Kadowaki, Ushizawa, Imai, Takahashi, Hirasawa, Tsujimoto and Hashizume2004), embryos (Potts et al., Reference Potts, Echternkamp, Smith and Reecy2003; Renard et al., Reference Renard, Sreenan and Hue2004b), endometrium (Renard et al., Reference Renard, Lewin, Yang, Hernandez, Sandra, Everts and Hue2004a), uterus and ovaries (Anderson et al., Reference Anderson, Finlayson and Archibald2004) and several pooled tissues (Smith et al., Reference Smith, Grosse, Freking, Roberts, Stone, Casas, Wray, White, Cho, Fahrenkrug, Bennett, Heaton, Laegreid, Rohrer, Chitko-McKown, Pertea, Holt, Karamycheva, Liang, Quackenbush and Keele2001). Several different bovine cDNA arrays were generated, specifically covering the transcriptome of, for example immune cells and tissues (Donaldson et al., Reference Donaldson, Vuocolo, Gray, Strandberg, Reverter, McWilliam, Wang, Byrne and Tellam2005; Jensen et al., Reference Jensen, Talbot, Paxton, Waddington and Glass2006) and other tissues of interest (Moody et al., Reference Moody, Rosa and Reecy2003). None of these molecular tools is specifically tailored for functional genomics studies of beef quality. Therefore, arrays have been constructed from cDNA libraries derived from bovine muscles or muscle and adipose tissue (Cho et al., Reference Cho, Han, Kang, Lee and Choi2002; Lehnert et al., Reference Lehnert, Wang and Byrne2004). The number of array elements printed on these tissue-specific arrays was generally lower than that derived from multiple tissues (Suchyta et al., Reference Suchyta, Sipkovsky, Kruska, Jeffers, McNulty, Coussens, Tempelman, Halgren, Saama, Bauman, Boisclair, Burton, Collier, DePeters, Ferris, Lucy, McGuire, Medrano, Overton, Smith, Smith, Sonstegard, Spain, Spiers, Yao and Coussens2003).
Researchers from the CRC for Cattle and Beef Quality in Australia constructed a bovine microarray of 9 600 elements comprising ca. 2 000 EST and ca. 7 300 anonymous cDNA clones from muscle and fat-derived cDNA libraries (Lehnert et al., Reference Lehnert, Wang and Byrne2004). The microarray also includes a collection of ‘candidate genes’ from the adipogenesis and protein turn-over literature. The main classes of genes represented on the microarray are coding for: contractile proteins (e.g. myosins, actins, etc.), extracellular matrix genes (e.g. collagens), mitochondrial enzymes (e.g. ATPases, cytochrome-c oxidase, etc.) and adipogenesis-related molecules or binding proteins (e.g. SCD, FABP4). These gene families are associated with the main cellular components of muscle tissue: the muscle fibres themselves, connective tissue and intramuscular fat islands. The bovine fat/muscle microarray has been used in a number of experiments to track gene expression changes in muscle tissue during nutritional treatments (Byrne et al., Reference Byrne, Wang, Lehnert, Harper, McWilliam, Bruce and Reverter2005; Lehnert et al., Reference Lehnert, Byrne, Reverter, Nattrass, Greenwood, Wang, Hudson and Harper2006) and in breed comparisons (Wang et al., Reference Wang, Byrne, Reverter, Harper, Taniguchi, McWilliam, Mannen, Oyama and Lehnert2005a and Reference Wang, Reverter, Mannen, Taniguchi, Harper, Oyama, Byrne, Oka, Tsuji and Lehnertb). The array has clear limitations for the discovery of regulatory mechanisms in muscle tissue development, as it was based on adult tissues only and therefore has very poor representation of probes for transcription factor genes and other regulatory factors.
One muscle specific library was prepared in France from selected various muscle types (oxidative or glycolytic) sampled from different bovine animals of various age (foetal and post-natal) and various breeds. Among the 1440 cDNAs characterised so far, 1019 from several gene families could be exploited: mitochondrial genes (133), genes encoding ribosomal proteins (100), contractile proteins, metabolic enzymes, etc. Finally, 353 cDNAs from this library and 75 cDNAs individually prepared by RT-PCR were considered as a first bovine muscle cDNA repertoire (Sudre et al., Reference Sudre, Leroux, Cassar-Malek and Hocquette2005b).
Pooled-tissue cDNA arrays
Tissue specific microarrays, particularly when they are based on adult tissues, have very clear limitations with respect to discovery of regulatory interactions, as they do not capture the whole transcriptome of the organism. cDNA microarrays with genome-wide representation have therefore been a goal of the scientific community since many EST collections became available and could be pooled (Table 3).
Table 3 Some bovine gene expression arrays suitable for beef quality studies (extracted, adapted and updated from Moody et al. (Reference Moody, Rosa and Reecy2003)). The reader should note that cDNA arrays are becoming superseded by oligo arrays and whole genome arrays

A bovine cDNA array (CattleArray 7600 from Pyxis Genomics) became available commercially in 2003. Since this array has been made with cDNAs derived from spleen and placenta, it may not be suitable to explore muscle biology and hence beef quality. A number of high-density cDNA arrays constructed for assessing genome-wide gene expression changes in cattle now include genes from muscle-specific libraries.
The NBFGC in the USA has selected 18 263 genes from the pooled tissues MARC 1–4 libraries for printing on a high-density microarray (Suchyta et al., Reference Suchyta, Sipkovsky, Kruska, Jeffers, McNulty, Coussens, Tempelman, Halgren, Saama, Bauman, Boisclair, Burton, Collier, DePeters, Ferris, Lucy, McGuire, Medrano, Overton, Smith, Smith, Sonstegard, Spain, Spiers, Yao and Coussens2003). Scientists at the Iowa University have followed the same strategy. They selected a set of 10 608 EST from MARC 1–4 libraries which include all the known genes and unknown, putative or hypothetical genes expressed in muscle, liver or adipose tissue. The selected genes include 4 484 known genes, 1 170 hypothetical proteins, 497 unknown and 4 364 not assigned genes. From a database of human genes expressed in muscle, liver and adipose tissue (http://telethon.bio.unipd.it/GETProfiles/Index.html), it was calculated that the collection contains 66.37%, 71.02% and 67.34% of the human genes expressed in these three tissues respectively. This new library was spotted with a set of negative and positive controls. This new molecular array has been called the BoviAnalyser. It is advertised as suitable to analyse cell proliferation and differentiation in response to, for instance, disease resistance or the ability of the animals to produce marbled beef.
Scientists at the Roslin Institute's ARK-Genomics Centre in the UK have constructed high-density microarrays based mainly on its collection of immune-specific EST (McGuire and Glass, Reference McGuire and Glass2005) and AgResearch has been using a 10 204 bovine cDNA microarray with broad genome coverage in bovine and ovine transcriptome studies (Diez-Tascón et al., Reference Diez-Tascón, Keane, Wilson, Zadissa, Hyndman, Baird, McEwan and Crawford2005).
French scientists have also prepared cDNA libraries from bovine mammary gland (Le Provost et al., Reference Le Provost, Lépingle and Martin1996) and embryos (Renard et al., Reference Renard, Sreenan and Hue2004b). Their strategy is now to combine high-quality cDNA clones from their different libraries (namely from muscle, mammary gland and embryo) in the same molecular tool for gene expression profiling in bovine tissues involved in reproduction and production traits. This was reported as successful (Bernard et al., Reference Bernard, Degrelle, Ollier, Campion, Cassar-Malek, Charpigny, Dhorne-Pollet, Hue, Hocquette, Le Provost, Leroux, Piumi, Roland, Uzbekova, Zalachas and Martin2005). In addition, a selection of 13 168 clones from the MARC 1–4 libraries (75%) and from INRA clones (25%) has been made to prepare an array suitable for many applications in cattle (http://sigenae.jouy.inra.fr/).
While cDNA libraries and cDNA arrays have allowed the bovine research communities to make rapid progress in transcriptomics, this technology platform is currently becoming superseded by oligo arrays and whole genome arrays.
Oligo arrays
Microarrays of synthetic oligonucleotide probes are becoming increasingly popular for transcriptomic studies, including for bovine (Table 3). The power of this approach lies largely in the opportunity to tap into the bovine genome and EST sequencing effort without the need to own libraries that contain probes of interest. Oligo arrays avoid some of the technical pitfalls of cDNA arrays, namely their lack of reliability and spot variability. The reliance on microbiological libraries of clones and their inherent contamination and handling risks has caused problems with reliability for many practitioners. The majority of the variability of microarray spots on cDNA arrays is due to the non-uniform performance of PCR reactions in probe generation. Oligo arrays are therefore of much better quality than cDNA arrays, even in heterologous systems. For instance, muscle-specific oligo arrays initially prepared for studies in mice and humans were shown to be well adapted to muscle gene expression studies in cattle (Bernard et al., Reference Bernard, Cassar-Malek, Dubroeucq, Renand and Hocquette2006b). Many bovine-specific oligo arrays have also been developed for transcriptomic studies in cattle (Table 3).
Operon technologies, Inc. market a bovine oligo probe set for microarray printing. It is derived from TIGR assemblies of bovine EST available in 2004 and contains 70mer probes (about 8 300 bovine genes with a known sequence with a hit to a known human, mouse, or cattle transcript). A bovine long oligo array project has been developed among a consortium of researchers at Texas A&M University, University of Missouri, Iowa State University, and the University of Minnesota. GenBank sequences are being analysed to remove duplicated sequences, vector sequences and other artefacts of the cloning process. This array was made available to the international community in 2006 (Elsik et al., Reference Elsik, Antoniou, Fahrenkrug, Reecy, Wolfinger and Taylor2006). Another oligo array project is currently developed between the University of Illinois, the University of Connecticut and INRA in France.
Affymetrix markets a high-density microarray, the GeneChip® Bovine Genome Array. It is representative of the publicly known, high-quality bovine sequences that were available in March 2004. It contains 24 027 probes. It is advertised to be suitable for studies related to disease resistance, meat and dairy production as well as stress tolerance.
Applications of functional genomics
Numerous results from transcriptomic studies have been obtained so far in bovine health, normal physiology or pathology. Gene expression profiling has been used to document changes in gene expression for example following infection by pathological organisms (Meade et al., Reference Meade, Gormley, Park, Fitzsimons, Rosa, Costello, Keane, Coussens and MacHugh2006), during the metabolic changes imposed by lactation in dairy cows (Loor et al., Reference Loor, Dann, Everts, Oliveira, Green, Guretzky, Rodriguez-Zas, Lewin and Drackley2005), in cloned bovine embryos (Smith et al., Reference Smith, Everts, Tian, Du, Sung, Rodriguez-Zas, Jeong, Renard, Lewin and Yang2005; Somers et al., Reference Somers, Smith, Donnison, Wells, Henderson, McLeay and Pfeffer2006) and in various other models (reviewed by Henderson et al., Reference Henderson, Thomas and Da2005 and by Hocquette et al., Reference Hocquette, Cassar-Malek, Listrat, Picard, Hocquette and Gigli2005).
It has been forecast that the identification of differentially expressed genes may be of high importance for muscle growth and development (Reecy et al., Reference Reecy, Spurlock and Stahl2006) and also for both genetic and physiological studies related to beef quality (Eggen and Hocquette, Reference Eggen and Hocquette2004). Despite this enthusiasm, some pitfalls need to be avoided. There are several reasons why the differentially expressed genes may not necessarily be the causal genes: (i) the lack of information collected on low-expressed genes (such as transcription factors and cellular regulators) is a limitation of the technology, (ii) muscle tissue is a complex mixture of cell types (myofibres, connective tissue fibroblasts, endothelial cells and adipocytes) and array studies can be simply affected by proportional changes in all these cell populations. Efforts to characterise the histological phenotype of the tissue alongside the gene expression phenotype will go some way towards addressing these issues. Notwithstanding these limitations, gene and protein expression profiling approaches have been directed at muscle of cattle (Table 4) and other livestock species, such as the pig (Lin and Hsu, Reference Lin and Hsu2005; Plastow et al., Reference Plastow, Carrion, Gil, Garcia-Regueiro, Furnols, Gispert, Oliver, Velarde, Guardia, Hortos, Rius, Sarraga, Diaz, Valero, Sosnicki, Klont, Dornan, Wilkinson, Evans, Sargent, Davey, Connolly, Houeix, Maltin, Hayes, Anandavijayan, Foury, Geverink, Cairns, Tilley, Mormede and Blott2005; Cagnazzo et al., Reference Cagnazzo, te Pas, Priem, de Wit, Pool, Davoli and Russo2006). Some recent insights into bovine muscle biology that have been obtained by cattle muscle profiling are described, as follows.
Table 4 Some examples of current initiatives in bovine genomics research applied to beef quality (extracted, adapted and updated from Henderson et al. (Reference Henderson, Thomas and Da2005) for US projects)
Comparison of gene expression between muscle types
The first aim when looking at gene expression in various muscle types is to get a better understanding of muscle characteristics, which determine meat quality traits across muscles since the major factor of variability of tenderness is the muscle type rather than cattle breed or animal type (heifer, young bulls or cull cows) (Dransfield et al., Reference Dransfield, Martin, Bauchart, Abouelkaram, Lepetit, Culioli, Jurie and Picard2003). Furthermore, some beef quality indicators, such as shear force, which is an indication of tenderness, are poorly correlated between different muscle types (Shackelford et al., Reference Shackelford, Wheeler and Koohmaraie1995). Therefore, understanding differences between muscle types is of major importance in meat science.
Differences between bovine muscle types were studied by using human macroarrays (Sudre et al., Reference Sudre, Leroux, Piétu, Cassar-Malek, Petit, Listrat, Auffray, Picard, Martin and Hocquette2003 and Reference Sudre, Cassar-Malek, Listrat, Ueda, Leroux, Jurie, Auffray, Renand, Martin and Hocquette2005a). As these arrays were initially prepared for other research objectives in humans, few differences were detected between muscles, however some genes of interest for muscle biology were revealed. Some of them were linked to the contractile (nebulin, actinin-associated LIM protein) or metabolic (ICDH β-subunit precursor) properties of muscles. Two interesting differentially expressed genes were LEU5 and Trip 15. LEU5 is a tumour suppressor gene associated with B-cell chronic lymphocytic leukemia. Trip 15 is a thyroid receptor interacting protein. They are probably involved in the regulation of muscle development. The differences between muscle types appeared to depend on the stage of development and the genetic types of animals (Sudre et al., Reference Sudre, Leroux, Piétu, Cassar-Malek, Petit, Listrat, Auffray, Picard, Martin and Hocquette2003 and Reference Sudre, Cassar-Malek, Listrat, Ueda, Leroux, Jurie, Auffray, Renand, Martin and Hocquette2005a).
Similar studies should also be performed to detect the specificity of intramuscular adipose tissue (which is important for beef flavour) in comparison to other fat tissues of the carcass undesirable for beef producers.
Gene expression according to the production system
The influence of two production systems (pasture v. maize-silage indoors) on muscle gene expression was studied in France. Transcriptomic analyses using a multi-tissue bovine cDNA repertoire were performed to compare gene expression profiling in rectus abdominis and semitendinosus muscles between both production groups. Variance analysis showed that the muscle type has an important effect on the expression level of genes. The effect of the production system was less marked. A list of the 30 most variable genes was established, of which 15 muscle genes were considered. Amongst them, the Selenoprotein W, which was found to be under-expressed on pasture, could be considered as an indicator of pasture-based system (Cassar-Malek et al., Reference Cassar-Malek, Bernard, Jurie, Barnola, Gentès, Dozias, Micol, Hocquette, Hocquette and Gigli2005a). However, whether variation of Selenoprotein W gene expression is linked to the feeding regime (grass v. maize-silage), or to the selenium status of the diet is still unclear.
Gene expression throughout development
Before the development of bovine cDNA arrays, human muscle-specific arrays were used to monitor gene expression throughout development (Sudre et al., Reference Sudre, Leroux, Piétu, Cassar-Malek, Petit, Listrat, Auffray, Picard, Martin and Hocquette2003). This allowed the identification of up to 110 genes differentially expressed from 110 days post-conception to 15 months of age after birth. Among these genes, 33% have unknown functions so far. Most of the differentially expressed genes are either up-regulated or down-regulated at 260 days of foetal development. This confirms the importance of the last three months of gestation in bovine muscle differentiation (Picard et al., Reference Picard, Lefaucheur, Berri and Duclos2002). Indeed, the number of muscle fibres is roughly fixed at two thirds of foetal development and muscle fibres differentiate before birth making cattle a relatively mature species at birth compared to other mammals. Not only did the macroarray experiment confirm this general view, but it also helped in the discovery of putative interesting genes regulated throughout development and involved in cell homeostasis (e.g. Sialyltransferase 8) or cell regulation (e.g. activin A, the thyroid receptor interacting protein 15) and in metabolic (e.g. oxoglutarate dehydrogenase) or contractile (e.g. nebulin) muscle properties (Sudre et al., Reference Sudre, Leroux, Piétu, Cassar-Malek, Petit, Listrat, Auffray, Picard, Martin and Hocquette2003). Further studies are needed to explore the biological functions of the novel identified genes.
Muscle gene expression profile associated with marbling
Some sectors of the beef industry are looking for gene markers that would identify animals that have a high propensity to accumulate intramuscular fat in order to produce tasty and tender meat. In addition to research on gene expression in perirenal adipose tissue before and after fattening (Oishi et al., Reference Oishi, Taniguchi, Nishimura, Yamada and Sasaki2000), research by Childs et al. (Reference Childs, Goad, Allan, Pomp, Krehbiel, Geisert, Morgan and Malayer2002) aimed to identify genes associated with intramuscular fat development. Differential-display polymerase chain reaction allowed the identification of a known gene (NAT1, a translational suppressor) by comparing muscle RNA from different finishing periods on high grain feeding. NAT1 was not previously suspected to play a role in fat accumulation. Putative functional genes were found to be differentially expressed (e.g. ATP citrate lyase) or, surprisingly, not differentially expressed (e.g. PPARγ) between extreme animals.
Australian and Japanese scientists undertook a microarray-based comparison of the longissimus muscle (LM) from Japanese Black (JB) and Holstein (HOL) cattle over an extended intensive feeding period to identify genes that may be involved in determining the unique ability of JB cattle to deposit intramuscular (IM) fat with lower melting temperature (Wang et al., Reference Wang, Byrne, Reverter, Harper, Taniguchi, McWilliam, Mannen, Oyama and Lehnert2005a). Three consecutive biopsies from LM tissue were taken and RNA isolated from three JB (Tajima strain) and three Holstein (HOL) animals at age 11 to 20 months. The gene expression changes in these samples were analysed using a bovine fat/muscle cDNA microarray (Lehnert et al., Reference Lehnert, Wang and Byrne2004). A mixed-ANOVA model was fitted to the intensity signals. Three hundred and thirty-five (4.8%) array elements were identified as differentially expressed genes in this breed × time comparison study. Genes preferentially expressed in JB are associated with monounsaturated fatty acid (MUFA) synthesis, fat deposition, adipogenesis development and muscle regulation, while examples of genes preferentially expressed in HOL come from functional classes involved in connective tissue and skeletal muscle development. The gene expression differences detected between the LM of the two breeds give important clues to the molecular basis for the unique features of the JB breed, such as the onset and rate of adipose tissue development, metabolic differences, and signalling pathways involved in converting carbohydrate to lipid during lipogenesis. These findings will impact on industry management strategies designed to manipulate intramuscular adipose development at different development stages to gain maximum return for beef products.
Gene and protein expression profile according to muscle growth potential
Muscle hypertrophy is of particular interest in beef meat production as it has a strong economic importance. High muscle development is associated with specific muscle characteristics in favour of meat tenderness but not of its flavour. To address the consequences of selection for increased growth rate and reduced fat content of the carcass, transcriptomic analyses of two muscles (rectus abdominis oxidative and semitendinosus glycolytic) from young bulls divergently selected on muscle growth potential were performed. The selection process in favour of muscle growth decreased oxidative muscle activity especially in rectus abdominis, which is the most oxidative muscle of both. Some genes were differentially expressed in rectus abdominis and/or semitendinosus between the two groups of bulls with high or low muscle growth potential. Many of them are involved in muscle structure (e.g. sarcosin, titin) or in cellular regulation (e.g. thyroid hormone receptor interacting protein 10, LEU5, an heat shock protein) and were more expressed in muscles from low muscle growth potential bulls compared to high muscle growth potential bulls (Sudre et al., Reference Sudre, Cassar-Malek, Listrat, Ueda, Leroux, Jurie, Auffray, Renand, Martin and Hocquette2005a).
Similarly, studies were conducted in double-muscled (DM) animals which are known to be characterised by a higher muscle mass and a lower fat content in the carcass and in the muscle tissue due to mutation in myostatin (a negative regulator of skeletal muscle growth which belongs to the transforming growth factor (TGF)-β superfamily.). One challenge is nowadays to elucidate the specific mechanisms, through which myostatin signals to inhibit the growth of skeletal muscle. About 20 genes were reported to be differentially expressed between DM and normal-muscled bovine embryos at 31 to 33 days of gestation. These differentially expressed genes fall into general classes including transcription factors (n = 5), genes involved in protein synthesis and degradation (n = 8), cell proliferation (n = 3), or altered metabolism (n = 4). Three of these genes were physically mapped to bovine chromosome 5 very close to a QTL for Warner Bratzler shear force (WBS) at day 14 post mortem (an indicator of the final toughness of beef). This may be a first step to explain the link between changes in meat tenderness associated with muscle hypertrophy (Potts et al., Reference Potts, Echternkamp, Smith and Reecy2003).
A recent study was conducted in order to compare the expression profile of muscle genes in the semitendinosus of double-muscled and non-double-muscled (NDM) 260-day-old foetuses using muscle-dedicated oligochips. The differential expression of several gene categories was found. Genes involved in slow contractile properties (e.g. TNNC1, TPM3, MYH7), extracellular matrix (e.g. collagen I and III, etc.) and ribosomal proteins (e.g. RPL3, RPL23, RPS24, RPS20) were found to be under-expressed in the DM foetuses. On the other hand, genes belonging to the regulation of cell cycle (e.g p21cip1, E2F1, CTBP1), DNA metabolism and the regulation of transcription (e.g. HMGB1, mcm6, HDAC4, MEF2A, MyoD), and the protease (e.g. furin, TIMP4) were found to be over-expressed in the DM foetuses compared to normal ones. Interestingly, the expression of three differentially expressed genes (Foxc2, SIX3 and ZFHX1B) was also found in DM cows, suggesting the putative involvement of these genes in the maintenance of muscle hypertrophy (Cassar-Malek et al., Reference Cassar-Malek, Passelaigue, Bernard, Gautier and Hocquette2005b). Further work is needed to understand their physiological implication in the development and modulation of muscle mass.
Similar transcriptomic studies were conducted in myostatin-knockout mice compared with wild-type mice using the Affymetrix GeneChip system. It was shown that myostatin may act upstream of Wnt pathway components, Wnt signalling being known to play a role in embryonic myogenesis. Several Wnt, including Wnt4, are capable of inducing some myogenic regulatory genes. Wnt4 is also capable of stimulating satellite cell proliferation. Based on gene expression results, a model was proposed in which Wnt4 is inhibited in the presence of myostatin, and activated in the absence of myostatin leading to stimulation of satellite cell proliferation. These results offer new insight into genes which may interact with myostatin to regulate skeletal muscle growth (Steelman et al., Reference Steelman, Recknor, Nettleton and Reecy2006).
To further understand how muscle hypertrophy is controlled, a proteomic analysis was developed to identify some markers of muscle mass. Two models of cattle with different origins of muscle hypertrophy: monogenic (DM Belgian Blue bulls) or polygenic (divergent lineages of Charolais bulls: with high (H) or low (L) muscle growth rate) were studied. Differential proteomic analysis of semitendinosus muscle was performed using two-dimensional gel electrophoresis (4 to 7 pH gradient in the first dimension and 11% SDS-PAGE in the second) followed by mass spectrometric analysis of interesting spots (MALDI-TOF) as described in Bouley et al. (Reference Bouley, Chambon and Picard2004a). Proteomic analysis revealed 13 proteins corresponding to 28 protein spots significantly altered between DM and NDM muscles (Bouley et al., Reference Bouley, Meunier, Chambon, De Smet, Hocquette and Picard2005). Among the proteins differentially expressed between the two genotypes, eight were related to contractile apparatus including myosin-binding protein H, several myosin light chains and troponin T (TnT) including slow TnT and fast TnT. Two proteins were involved in metabolic pathways, including phosphoglucomutase and the heart fatty acid-binding protein. Finally, three proteins were significantly altered including sarcosin, sarcoplasmic reticulum 53 kDa glycoprotein and p20. According to these data it seems that contractile properties are more modified than metabolic ones in DM muscles. Most of the modifications of protein expression are in favour of fast glycolytic properties of DM muscle. This is coherent with the data of the literature showing an orientation of muscle type towards the fast glycolytic type in case of muscle hypertrophy (reviewed by Hocquette et al. (Reference Hocquette, Ortigues-Marty, Pethick, Herpin and Fernandez1998)). A similar analysis was conducted on semitendinosus muscle from H and L young bulls. Interestingly, it revealed that the proteins differentially expressed in DM muscle were also differentially expressed in H muscle (Picard et al., Reference Picard, Bouley, Cassar-Malek, Bernard, Renand, Hocquette, Hocquette and Gigli2005). Among these proteins, eleven spots of fast TnT (fTnT) were analysed more precisely. The expression levels of five of them were significantly increased in DM and H muscles. The levels of expression of the other six were unchanged or decreased in DM and H muscles. These fTnT isoforms differ in terms of the presence of the alternative splicing region corresponding to mutually exclusive exons 16 and 17 (Bouley et al., Reference Bouley, Meunier, Chambon, De Smet, Hocquette and Picard2005, Muroya et al., Reference Muroya, Nakajima and Chikuni2003). All these data demonstrated that the TnT fast isoforms over-expressed in the muscle of high muscle growth potential (DM and H) contained the exon 16 and the others contain the exon 17. This could constitute a good protein marker of muscle hypertrophy. This higher expression of fTnT with exon 16 in muscles of high growth potential showing an over expression of fast contractile proteins is coherent with the data from the literature: indeed, Bucher et al. (Reference Bucher, De La Brousse and Emerson1989) showed a correlation between the expression of fTnT exon 16 and fast contractile status of muscle. Also, Jin et al. (Reference Jin, Wang and Ogut1998) found a higher proportion of fTnT cDNA encoding the mutually exclusive fTnT exon 16 in a fast glycolytic muscle (Pectoralis).
Gene and protein expression associated with tenderness
Transcriptomic and proteomic analysis of bovine muscle was also carried out in order to identify some markers of meat tenderness. For transcriptomic studies, samples of longissimus muscles from 14, 15- and 19-month-old Charolais bulls were analysed using oligo chips. Gene expression profiles were compared between high and low meat quality scores of tenderness, flavour and juiciness. Several genes were differentially expressed. Fourteen of them were highly correlated with flavour and juiciness, and one of them had a strong negative correlation with tenderness (Bernard et al., Reference Bernard, Cassar-Malek, Dubroeucq, Renand and Hocquette2006b). This gene called DNAJA1 was patented because it is responsible for up to 63% of the variability in meat tenderness (Bernard et al., Reference Bernard, Cassar-Malek, Hocquette and collaborators2006a) although these results have to be validated using a greater number of samples. DNAJA1 encodes a protein which is a member of the large heat shock 40kDa protein family. This protein is a co-chaperone of Hsc70 and could to play a role in protein import into mitochondria. Its involvement in beef tenderness remains unknown.
Similarly, samples of the semitendinosus muscle previously sorted according to different eating quality classes on the basis of the results of sensory analysis (Dransfield et al., Reference Dransfield, Martin, Bauchart, Abouelkaram, Lepetit, Culioli, Jurie and Picard2003) were analysed by proteomic approaches. These muscles had been taken from 15-month-old bulls of three French breeds: Charolaise, Limousine and Salers. For the Charolaise and Limousine breeds, five protein spots were significantly altered between two classes of tenderness (Bouley et al., Reference Bouley, Meunier, Culioli and Picard2004b). Myosin binding protein H, Acyl-coA-binding protein, parvalbumin and a non identified protein were over-expressed in muscles of the high tenderness class. Myosin regulatory light chain 2 was under-expressed in the high tenderness class. However, these changes were not observed for the hardy Salers breed in which other proteins were altered. Because these preliminary results show that the same indicators were found in both beef breeds, this indicates that in hardy breeds such as Salers the determinism of tenderness might be different. Further analyses are in progress to complete these results on a more consequent number of samples.
Conclusion
Genomic research in cattle has to be considered in its different aspects. From the technical point of view, cattle genomics cannot be considered in isolation but will be heavily influenced by the general progress in mammalian genomics. From the scientific point of view, we must recognise that some interesting results have been obtained so far with regard to linking genomics results with aspects of beef quality. These two aspects will be dramatically reshaped with the availability of the next draft of the bovine genome sequence. The last point of view, which is of tremendous importance for economic considerations, is the prospect of practical applications of genome-based technologies to the beef industry.
From the technical point of view, genomic research of cattle is somewhat lagging behind that of model organisms despite a great deal of technical research which was recently done in cattle. Many bovine cDNA libraries have been constructed, and various bovine arrays were prepared either from cDNA libraries or from oligonucleotide probe sets designed from publicly available bovine sequences. Basic improvements in genomics using model organisms are likely to produce new tools and knowledge useful for genomics in cattle. On the other hand, the involvement of statisticians experienced in genetic statistics in cattle genomic research teams has led to some innovative developments in basic statistic methods of general interest to the microarray community. The bovine genome sequencing is forecast to be very useful for mammalian biology and health by comparing the bovine genome sequence to that of other species.
From the scientific point of view, this review has reported differentially expressed genes which were detected during bovine muscle development, between different breeds or different genotypes, between different nutritional levels or between different types of diet. A few markers of muscle hypertrophy or of beef tenderness were also identified at the mRNA or protein levels. The challenge is now to check the relevance of those key genes or proteins with the aim to use them in the near future to improve breeding or rearing strategies. The muscle tissue is however a complex tissue with different cell types (multi-nucleated fibres, adipocytes, etc.) in various proportions between muscles or even within the same muscle. Another challenge is to investigate which cell population is responsible for any change in gene expression.
From the economic point of view, the importance of genomics in modern agricultural practice is likely to increase. The education of producers is therefore a key issue in the adaptation of genomic research results to agricultural practice. The application of genomics and proteomics research has the potential to generate tools for beef producers that will help them to improve beef quality or to produce differentiated products. Over the longer term, these tools have the potential to change production methods in cattle breeding, husbandry and nutrition. In order to ensure efficient adoption of the outcomes of genomic research, ethical, legal, environmental, consumer and any societal concerns with the technology have to be addressed. A first success in that direction is that private partners representative of the beef industry are working together with public institutions to allow the development of the genomic research.




