Implications
Meta-omics is playing an increasingly important role in defining the ecology of the rumen ecosystem and its responses to changes in diet. Metatranscriptomics is shedding light on rumen function and gaging the contribution of microbiota to important ruminal metabolic processes such as carbohydrate and protein digestion. Emerging long-read sequencing technologies are sure to advance our present understanding of interrelationships among microbes and their genes. Application of these technologies in a manner that focuses on host – rumen microbiome interactions is the key to further advancing our understanding of this fascinating ecosystem.
The rumen and the rumen microbiome
Ruminants evolved approximately 50 million years ago and are among the most widely adapted large mammals on earth, with an estimated 200 species inhabiting environments from the arctic to the tropics (Hackmann and Spain, Reference Hackmann, Ngugi, Firkins and Tao2010). They are unique among livestock because they can efficiently utilize forages, food by-products and non-protein nitrogen to produce milk and meat, thereby avoiding plant materials more suitable for human consumption. Like all mammalian herbivores, ruminants do not produce cellulolytic or hemi-cellulolytic enzymes to degrade ingested plant material. Instead, they rely on symbiotic associations with bacteria, fungi and protozoa within the rumen to perform this function. The rumen microbiome comprises bacteria (up to 1011 cells/ml), protozoa (104 to 106 cells/ml), fungi (103 to 106 zoospores/ml), methanogens (106 cells/ml) and bacteriophages (107 to 1010 particles/ml) (Morgavi et al., Reference Morgavi, Kelly, Janssen and Attwood2013). These microbial symbionts are highly specialized in degrading lignocellulosic biomass, and the ruminant host is dependent on the array of enzymes produced by the microbial community to convert complex fibrous substrates into fermentable saccharides. Ultimately, sugars released from plant material are fermented by rumen bacteria and converted primarily into the volatile fatty acids (VFAs), acetate, propionate and butyrate. These VFAs along with microbial protein are utilized by the ruminant for maintenance, growth and lactation (Morgavi et al., Reference Morgavi, Kelly, Janssen and Attwood2013).
Composition of the rumen microbiome
Great efforts have been made to understand the composition of the rumen microbial community and how it changes in response to a variety of selective pressures (Huws et al., Reference Huws, Mayorga, Theodorou, Kim, Cookson, Newbold and Kingston-Smith2018). Initial studies involved culturing rumen bacteria directly associated with the digestion of cellulose/hemicellulose and generated a substantial body of knowledge on the biology of the principal cellulolytic bacteria, Fibrobacter succinogenes, Ruminococcus albus, Ruminococcus flavefaciens and Butyrivibrio fibrisolvens (Morgavi et al., Reference Morgavi, Kelly, Janssen and Attwood2013) and the rumen fungi (Edwards et al., Reference Edwards, Huws, Kim and Kingston-Smith2007). Far less is known about the rumen virome, and only recently has genomics shed light on the possible role of viruses within the rumen microbiota (Anderson et al., Reference Anderson, Sullivan and Fernando2017; Gilbert et al., Reference Gilbert, Kelly, Altemann, Leahy, Minchin, Ouwerkerk and Klieve2017). Although bacteria are better characterized, only 15% of the total species have been cultured in the laboratory (Morgavi et al., Reference Morgavi, Kelly, Janssen and Attwood2013; Creevey et al., Reference Creevey, Kelly, Henderson and Leahy2014). Currently, there are at least 70 rumen bacterial species available in pure culture from public repositories (Creevey et al., Reference Creevey, Kelly, Henderson and Leahy2014). Recent large-scale projects such as the Global Rumen Census (Henderson et al., Reference Henderson, Cox, Ganesh, Jonker, Young and Janssen2015) and the Rumen Microbial Genomics Network Hungate1000 project (Seshadri et al., Reference Seshadri, Leahy, Attwood, Teh, Lambie, Cookson, Eloe-Fadrosh, Pavlopoulos, Hadjithomas, Varghese, Paez-Espino, Perry, Henderson, Creevey, Terrapon, Lapebie, Drula, Lombard, Rubin, Kyrpides, Henrissat, Woyke, Ivanova and Kelly2018) have enhanced our understanding of the rumen microbial ecosystem and its role in fibre digestion. The Hungate1000 project sought to produce a reference set of rumen microbial genome sequences from cultivated rumen bacteria, methanogens, anaerobic fungi and ciliate protozoa. The project successfully generated a catalogue of 480 bacterial and 21 archaeal genomes but failed to generate complete genomes for either fungi or protozoa. The AT-rich nature of these eukaryotic genomes presents significant assembly challenges, but progress is being made in characterizing the functional genome of anaerobic fungi (Solomon et al., Reference Solomon, Haitjema, Henske, Gilmore, Borges-Rivera, Lipzen, Brewer, Purvine, Wright, Theodorou, Grigoriev, Regev, Thompson and O’malley2016; Haitjema et al., Reference Haitjema, Gilmore, Henski, Solomon, de Groot, Mondo, Salamov, LaButti, Zhao, Chiniquy, Barry, Brewer, Purvine, Wright, Hainaut, Boxma, van Alen, Hackstein, Henrissat, Baker, Grigoriev and O’Malley2017; Gruninger et al., Reference Gruninger, Nguyen, Reid, Yanke, Wang, Abott, Tsang and McAllister2018).
In addition to studies involving pure cultures, the application of cultivation-independent methods including metagenomics, metatranscriptomics and metaproteomics has greatly enhanced the scientific communities’ understanding of the structure and function of the rumen microbial ecosystem (Ribeiro et al., Reference Ribeiro, Gruninger, Badhan and McAllister2016). These cultivation-independent studies suggest that many of the microbes and carbohydrate-active enzymes (CAZymes) involved in lignocellulose digestion in the rumen are uncharacterized (Hess et al., Reference Hess, Sczyrba, Egan, Kim, Chokhawala, Schroth, Luo, Clark, Chen, Zhang, Mackie, Pennacchio, Tringe, Visel, Woyke, Wang and Rubin2011). One tool that has seen success in exploring the functions of these uncharacterized microbes is the assembly of bacterial and archaeal genomes from metagenomic sequence data from the rumen (Svartström et al., Reference Svartström, Alneberg, Terrapon, Lombard, de Bruijn, Malmsten, Dalin, El Muller, Shah and Wilmes2017; Stewart et al., Reference Stewart, Auffret, Warr, Wiser, Press, Langford, Liachko, Snelling, Dewhust, Walker, Roche and Watson2018a). This method has been rapidly expanding the database of rumen microbial genome sequences. Despite the utility of this approach, it is imperative that the function of these genomes be confirmed through culturing and metabolomic confirmation. Continued efforts at exploring the microbial diversity present in the rumen could prove to be a valuable source of novel enzymatic activities and metabolic pathways that have applications for improving the feed conversion of ruminants and industrial biomass conversion with a focus on recalcitrant plant cell walls. Differentiating dietary conditions where this diversity contributes to improved feed efficiency from when it does not could be the key to obtaining consistent improvements in ruminal feed efficiency.
The Global Rumen Census described the composition of the rumen and camelid foregut bacterial and archaeal communities (i.e., 742 samples from 32 animal species across 35 countries) and how they were influenced by diet, host species and geography (Henderson et al., Reference Henderson, Cox, Ganesh, Jonker, Young and Janssen2015). This project found that the rumen microbiome was significantly influenced by host species and diet, with diet being the most influential factor. The taxonomic identity of the major microbial genera was similar, but large differences in the relative abundance of these bacteria were observed. This study highlights the adaptability of the rumen microbiota and agrees with an earlier work that found that the majority of the variation in the gut microbial community of mammals can be attributed to differences in diet (Ley et al., Reference Levy and Jami2008). Variation in the physical and chemical composition of the diet is believed to provide unique ecological niches that favour the selection of specific microbes (Henderson et al., Reference Henderson, Cox, Ganesh, Jonker, Young and Janssen2015). The rumen microbial community works in a complex and interlinked manner. Changes in the diet of the host promotes changes in ruminal microbial metabolism, altering the production of VFAs and methane, and ultimately impacting meat and milk production (de Menezes et al., Reference Deusch, Camarinha-Silva, Conrad, Beifuss, Rodehutscord and Seifert2011).
The Global Rumen Census also found that the rumen microbial ecosystem is dominated by a core community composed of poorly characterised microbes (Henderson et al., Reference Henderson, Cox, Ganesh, Jonker, Young and Janssen2015). The community was highly diverse at the level of the operational taxonomic unit (OTU) but, 89.4% of all sequences could be classified to just 30 bacterial groups that were found in over 90% of the samples (Henderson et al., Reference Henderson, Cox, Ganesh, Jonker, Young and Janssen2015). The core bacterial OTUs belonging to Prevotella, Butyrivibrio, Ruminococcus and the unclassified Lachnospiraceae, Ruminococcaceae, Bacteroidales and Clostridiales were the most abundant bacterial groups in the rumen (67.1% of all bacterial sequence data). A greater abundance of unclassified Bacteroidales and Ruminococcaceae were observed in the rumen of animals fed high-forage diets, and greater abundance of Prevotella and unclassified Succinivibrionaceae were observed in the rumen of animals fed diets containing concentrate. The common rumen bacteria Prevotella ruminicola, P. brevis, P. bryantii and P. albensis tended to be more abundant in high-concentrate diets, and Fibrobacter abundance was higher in forage-fed cattle (Henderson et al., Reference Henderson, Cox, Ganesh, Jonker, Young and Janssen2015). In addition, higher microbial diversity is also observed in forage diets as compared to high-concentrate diets (Tapio et al., Reference Tapio, Fischer, Blasco, Tapio, Wallace, Bayat, Ventto, Kahala, Negussie and Shingfield2017), a finding that likely reflects the greater complexity of carbohydrates and possibly functional conditions (i.e., pH, rate of passage, osmolality) in ruminants fed forage-based diets.
Recent efforts using network analysis to examine the metabolic function of rumen microbial communities found that distinct taxa can have similar metabolic networks and perform similar metabolic processes (Taxis et al., Reference Taxis, Wolff, Gregg, Minton, Zhang, Dai, Schnabel, Taylor, Kerley, Pires, Lamberson and Conant2015). This redundancy was postulated to be due to similarity in the metabolic inputs and outputs between different taxa. The significance of the high metabolic redundancy present in the rumen is not known, but it has been suggested that it plays a role in carbohydrate digestion (Weimer, Reference Wenner, Wagner and Firkins2015). All -omics-based methods rely heavily on well-annotated databases to assign function to genes/proteins. A recent study of the metabolic pathways present in the genomes of a number of rumen bacteria identified several non-standard metabolic pathways that are not adequately represented in current databases (Hackmann et al., Reference Hackmann and Spain2017). Hackmann and colleagues found that the recognized pathways for metabolizing pentose and hexose sugars to short-chain fatty acids do not adequately explain the fermentation products generated by a wide range of rumen bacteria (Hackmann et al., Reference Hackmann and Spain2017). They found that 44% of these bacteria encoded atypical metabolic pathways and identified several that are completely novel. This finding underscores the need to continue culturing rumen microbes to facilitate the development of a well-annotated, diverse sequence database that accurately represents the biological processes present in the rumen.
Beyond bacteria: methanogenic archaea and eukaryotic members of the rumen microbiome community
The rumen is also inhabited by archaea, fungi and protists. The archaea found in the rumen are exclusively methanogens, and these microbes primarily produce methane by the reduction of CO2 with H2 (Wang et al., Reference Wang, Elekwachi, Jiao, Wang, Tang, Zhou, Tan and Forster2017). The archaeal community is significantly less diverse than the bacterial community and was found to be similar across all regions of the world (Henderson et al., Reference Henderson, Cox, Ganesh, Jonker, Young and Janssen2015). Methanobrevibacter gottschalkii and Methanobrevibacter ruminantium were the most abundant archaea and were found in almost all samples. The dominance of archaea within the genus Methanobrevibacter is consistent with the majority of previous studies examining the methanogen community in the rumen (Janssen and Kirs, Reference Janssen and Kirs2008; Wang et al., Reference Wang, Elekwachi, Jiao, Wang, Tang, Zhou, Tan and Forster2017). Together with Methanosphaera sp. and two Methanomassiliicoccaceae-affiliated groups, five genera of methanogenic archaea comprised 89.2% of the community (Henderson et al., Reference Henderson, Cox, Ganesh, Jonker, Young and Janssen2015). The recently named order Methanomassiliicoccales is phylogenetically related to Thermoplasmatales and has been referred to by a number of names including ’rumen cluster C‘, ‘Thermoplasmatales affiliated lineage C’, or ‘order Methanoplasmatales’ (Borrel et al., Reference Borrel, Parisot, Harris, Peyretaillade, Gaci, Tottey, Bardot, Raymann, Gribaldo, Peyret, O’Toole and Brugere2014; Wang et al., Reference Wang, Elekwachi, Jiao, Wang, Tang, Zhou, Tan and Forster2017). This group of methanogenic archaea is commonly found in the rumen, and in some ruminants have been found to make up a significant proportion of the archaeal community (Wang et al., Reference Wang, Xu, Kong, Yang, Wu, Mishra and Li2016b).
Much less is known about the ecology of the eukaryotic component of the rumen microbial community, namely the rumen fungi (Phylum Neocallimastigomycota) and the ciliated rumen protists (Groups Isotricha and Eudiplodinium). The anaerobic fungi play a key role in the initial colonization and degradation of the plant cell wall through the concerted action of carbohydrate-active enzymes and physical penetration of the plant cell wall by hyphae (Edwards et al., Reference Edwards, Huws, Kim and Kingston-Smith2007). There are currently 11 cultured anaerobic fungi; however, metagenomic studies targeting the Internal transcribed spacer 1 (ITS-1) amplicon have revealed much greater taxonomic diversity in the rumen fungi with at least seven more phylogenetically distinct clades that are not represented by cultured isolates (Liggenstoffer et al., Reference Liggenstoffer, Youssef, Couger and Elshahed2010; Koetschan et al., Reference Koetschan, Kittelmann, Lu, Al-Halbouni, Jarvis, Müller, Wolf and Janssen2014; Tapio et al., Reference Tapio, Fischer, Blasco, Tapio, Wallace, Bayat, Ventto, Kahala, Negussie and Shingfield2017). Rumen fungi are associated with forage-based diets, and their abundance decreases rapidly upon the addition of concentrate to the diet (Belanche et al., Reference Belanche, de la Fuente and Newbold2012). In 2011, the first metatranscriptomic study of the rumen sequenced transcripts from the eukaryotic members of the rumen microbial community (Qi et al., Reference Qi, Wang, O’Toole, Barboza, Ungerfeld, Leigh, Selinger, Butler, Tsang and McAllister2011). In this seminal study, Qi and colleagues found that rumen eukaryotes contributed a significant portion of diverse cellulases, hemicellulases and esterases and that these proteins showed low levels of sequence identity to characterized proteins. In addition, many of these enzymes belonged to carbohydrate-active enzyme families that are not found in bacteria (Qi et al., Reference Qi, Wang, O’Toole, Barboza, Ungerfeld, Leigh, Selinger, Butler, Tsang and McAllister2011).
Advances in sequencing technology and bioinformatics tools have enabled researchers to sequence the genomes of four species of Neocallimastigomycota (Youseff et al., Reference Youssef, Couger, Struchtemeyer, Liggenstoffer, Prade, Najar, Atiyeh, Wilkins and Elshahed2013; Haitjema et al., Reference Haitjema, Gilmore, Henski, Solomon, de Groot, Mondo, Salamov, LaButti, Zhao, Chiniquy, Barry, Brewer, Purvine, Wright, Hainaut, Boxma, van Alen, Hackstein, Henrissat, Baker, Grigoriev and O’Malley2017). All of these genome assemblies are fragmented to varying degrees. Despite this challenge, these genome sequences have revealed valuable insight into the evolution of anaerobic fungi and the adaptations they have acquired to survive in a competitive anaerobic environment. Some of these adaptations are the presence of tetrahymanol in the plasma membrane instead of ergesterol, pyruvate metabolism occurring via mixed acid fermentation (Youssef et al., Reference Youssef, Couger, Struchtemeyer, Liggenstoffer, Prade, Najar, Atiyeh, Wilkins and Elshahed2013) and the use of hydrogenosomes for ATP generation (Yarlett et al., Reference Yarlett, Orpin, Munn, Yarlett and Greenwood1986). The existence of cellulosomes in Neocallimastigomycota has long been proposed, but until recently the identity of the scaffoldin protein was not known. Using a combination of genomics and proteomics, the scaffoldin in Neocallimastigomycota was identified, and the dockerin-scaffoldin interaction has been examined (Haitjema et al., Reference Haitjema, Gilmore, Henski, Solomon, de Groot, Mondo, Salamov, LaButti, Zhao, Chiniquy, Barry, Brewer, Purvine, Wright, Hainaut, Boxma, van Alen, Hackstein, Henrissat, Baker, Grigoriev and O’Malley2017). Unlike the high-specificity dockerin-scaffoldin interaction in bacterial cellulosomes, the anaerobic fungal scaffoldin showed cross-reactivity between dockerin-containing proteins from multiple species of anaerobic fungi. This feature of the fungal cellulosome may be important for the co-existence of multiple species of fungi within the rumen (Haitjema et al., Reference Haitjema, Gilmore, Henski, Solomon, de Groot, Mondo, Salamov, LaButti, Zhao, Chiniquy, Barry, Brewer, Purvine, Wright, Hainaut, Boxma, van Alen, Hackstein, Henrissat, Baker, Grigoriev and O’Malley2017). More recently, a transcriptomic analysis of carbohydrate metabolism in diverse species of anaerobic fungi found that the carbohydrate-active enzymes expressed by several species of rumen fungi may preferentially target different carbohydrate components within the plant cell wall (Gruninger et al., Reference Gruninger, Nguyen, Reid, Yanke, Wang, Abott, Tsang and McAllister2018). It was suggested that this is another mechanism that may decrease the direct competition for resources and facilitate the co-existence of multiple species within the rumen (Gruninger et al., Reference Gruninger, Nguyen, Reid, Yanke, Wang, Abott, Tsang and McAllister2018). These genomic (Youssef et al., Reference Youssef, Couger, Struchtemeyer, Liggenstoffer, Prade, Najar, Atiyeh, Wilkins and Elshahed2013; Haitjema et al., Reference Haitjema, Gilmore, Henski, Solomon, de Groot, Mondo, Salamov, LaButti, Zhao, Chiniquy, Barry, Brewer, Purvine, Wright, Hainaut, Boxma, van Alen, Hackstein, Henrissat, Baker, Grigoriev and O’Malley2017) and transcriptomic (Solomon et al., Reference Solomon, Haitjema, Henske, Gilmore, Borges-Rivera, Lipzen, Brewer, Purvine, Wright, Theodorou, Grigoriev, Regev, Thompson and O’malley2016; Henske et al., Reference Henske, Gilmore, Knop, Cunningham, Sexton, Smallwood, Shutthanandan, Evans, Theodorou and O’Malley2017; Gruninger et al., Reference Gruninger, Nguyen, Reid, Yanke, Wang, Abott, Tsang and McAllister2018) studies of rumen fungi have yielded valuable insight into the biology of these microbes and found that a core set of genes and diverse array of plant cell well–degrading enzymes have been maintained throughout the evolution of the phylum Neocallimastigomycota. These features likely facilitate the survival of these microbes in the competitive rumen environment.
It has been proposed that ciliate protozoa may account for up to 50% of the rumen microbial biomass (Hungate et al., Reference Hungate, Reichl and Prins1971), but more recent studies that have measured protozoal volume using video microscopy have suggested that it may be far lower (Wenner et al., Reference Weimer2018). The role of protozoa within the rumen microbiome is considered controversial (Newbold et al., Reference Newbold, de la Fuente, Belanche, Ramos-Morales and McEwan2015). Studies with protozoa-free ruminants have demonstrated that they are not absolutely essential for rumen function. Protozoa are highly proteolytic and through bacterial predation are responsible for the turnover of a large portion of the microbial protein within the rumen (Figure 1). Protozoa also contribute to the degradation of feed protein and are associated with higher ruminal concentration of ammonia nitrogen. A myriad of protozoa eradication or removal strategies have been developed because of the association these microbes have with decreased efficiency of rumen microbial protein synthesis, and increased methane emissions (Guyader et al., Reference Guyader, Eugene, Noziere, Morgavi, Doreau and Martin2014; Newbold et al., Reference Newbold, de la Fuente, Belanche, Ramos-Morales and McEwan2015). The rumen ciliates associated with methane production can harbour both epi- and endo-symbiotic methanogens which derive hydrogen from hydrogenosome organelles within these protozoa (Ellis et al., Reference Ellis, Williams and Lloyd1994). Eliminating protozoa from the rumen has proven difficult, and an effective treatment or feed additive is not currently commercially available. Contrarily, it has also been shown that protozoa can also have positive effects on ruminal feed digestion (Huws et al., Reference Huws, Mayorga, Theodorou, Kim, Cookson, Newbold and Kingston-Smith2018). Furthermore, protozoa can rapidly engulf starch granules when animals are fed high-grain diets, competing with ruminal amylolytic bacteria for substrate and reducing the rate of starch fermentation. Such events can slow down the rate of starch fermentation, possibly reducing the risk of ruminal acidosis (Owens et al., Reference Owens, Secrist, Hill and Gill1998; Newbold et al., Reference Newbold, de la Fuente, Belanche, Ramos-Morales and McEwan2015). In high-forage diets rumen protozoa have been estimated to account for 17% to 21% of total fibre degradation (Dijkstra and Tamminga, Reference Dijkstra and Tamminga1995). Studies in defaunated sheep fed forage diets showed that fibre digestibility declined by 14% to 17% (Belanche et al., Reference Belanche, Abecia, Holtrop, Guada, Castrillo, de la Fuente and Balcells2011, Reference Belanche, Doreau, Edwards, Moorby, Pinloche and Newbold2015). Protozoa also directly aid rumen fibre degradation through their involvement in the initial stages of fibre colonization and production of glycosyl hydrolases (GHs), as well as indirectly, through their consumption of low concentrations of O2 that can enter the rumen and destabilize ruminal metabolism (Hillman et al., Reference Hillman, Lloyd and Williams1985; Ellis et al., Reference Ellis, Lindmark, Williams and Lloyd1989; Findley et al., Reference Findley, Mormile, Sommer-Hurley, Zhang, Tipton, Arnett, Porter, Kerley and Stacey2011; Newbold et al., Reference Newbold, de la Fuente, Belanche, Ramos-Morales and McEwan2015). Protozoa also have complex interactions with other rumen microbes: a meta-analysis of defaunated ruminants indicated that they possessed fewer fibrolytic microbes, including anaerobic fungi (decreasing by 92%), R. albus (decreasing by 34%) and R. flavefaciens (decreasing by 22%) (Newbold et al., Reference Newbold, de la Fuente, Belanche, Ramos-Morales and McEwan2015). However, the lack of sequenced genomes for rumen protozoa has made advancements in the understanding of their role in ruminal degradation of carbohydrates and proteins using ’meta-omic’ approaches difficult (Comtet-Marre et al., Reference Comtet-Marre, Parisot, Lepercq, Chaucheyras-Durand, Mosoni, Peyretaillade, Bayat, Shingfield, Peyret and Forano2017).

Figure 1 Microbes associated with the rumen protozoa Polyplastron and Metadinium. Note that microbiomes associated with feed particles are also visible in the lower- and upper-left regions of the image with Polyplastron. Microbiomes can consist of complex or simple communities as illustrated by the almost-exclusive colonization of the outer surface of Metadinium by methanogens. Protozoa also perform a number of important functions within the rumen microbiome community. Blue bar = 10 µm. Protozoal samples were stained and visualized as described by Valle et al. (Reference Valle, Henderson, Jansen, Cox, Alexander and McAllister2015).
Studying the rumen microbiome with -omics approaches
Until recently, the rumen microbiome was primarily studied using culture-based or classical molecular techniques, including restriction fragment length polymorphism (RFLP), denaturing gradient gel electrophoresis (DGGE) and ribosomal RNA clone libraries. These molecular techniques have become largely obsolete, as the complexity of the rumen microbiome makes approaches such as metagenomics, metatranscriptomics and metaproteomics more meaningful for the study of the rumen microbiome. Amplicon-based metagenomic studies, also known as metataxonomics, sequence regions of marker genes such as the 16S rRNA, methyl coenzyme M reductase A (mcrA), 18S rRNA or ITS-1 that can be used to describe the bacterial, archaeal, protozoal and fungal populations, respectively (Li et al., Reference Li, Henderson, Sun, Cox, Janssen and Guan2016). These studies provide information on the composition of the microbial community, but little information about their function (Dai et al., Reference D’Amore, Ijaz, Schirmer, Kenny, Gregory, Darby, Shakya, Podar, Quince and Hall2015; Shinkai et al., Reference Shinkai, Mitsumori, Sofyan, Kanamori, Sasaki, Katayose and Takenaka2016; Comtet-Marre et al., Reference Comtet-Marre, Parisot, Lepercq, Chaucheyras-Durand, Mosoni, Peyretaillade, Bayat, Shingfield, Peyret and Forano2017). In contrast, shotgun metagenomics, metatranscriptomics and metaproteomics can provide a snapshot of both the taxonomic composition and the metabolic activity of a rumen microbial community under a particular set of conditions that dictate function. Metagenome-based approaches tend to be biased towards numerically abundant genes harboured by the most abundant microbial species. Increasingly, shotgun metagenomics is being used to reconstruct whole genomes of uncultured rumen microbes, providing novel insight into the biological mechanisms they have evolved to let them thrive in the rumen (Hess et al., Reference Hess, Sczyrba, Egan, Kim, Chokhawala, Schroth, Luo, Clark, Chen, Zhang, Mackie, Pennacchio, Tringe, Visel, Woyke, Wang and Rubin2011; Svartström et al., Reference Svartström, Alneberg, Terrapon, Lombard, de Bruijn, Malmsten, Dalin, El Muller, Shah and Wilmes2017).
Trying to understand a highly complex community such as the rumen using a single approach leads to the development of an incomplete picture. To address this limitation, a greater number of researchers are using a multi-omics approach combining metagenomics, metatranscriptomics, metaproteomics and metabolomics to study the rumen (Shinkai et al., Reference Shinkai, Mitsumori, Sofyan, Kanamori, Sasaki, Katayose and Takenaka2016; Comtet-Marre et al., Reference Comtet-Marre, Parisot, Lepercq, Chaucheyras-Durand, Mosoni, Peyretaillade, Bayat, Shingfield, Peyret and Forano2017; Deusch et al., Reference Dias, Marcondes, Noronha, Resende, Machado, Mantovani, Dill-McFarland and Suen2017). In the study of the rumen, combining the results of metatranscriptomics and metagenomics has provided novel insights into the specific microbes active in lignocellulose digestion (Qi et al., Reference Qi, Wang, O’Toole, Barboza, Ungerfeld, Leigh, Selinger, Butler, Tsang and McAllister2011; Dai et al., Reference D’Amore, Ijaz, Schirmer, Kenny, Gregory, Darby, Shakya, Podar, Quince and Hall2015; Li and Guan, Reference Li and Guan2017). A metatranscriptome study of dairy cows on a forage-based diet found that the most-expressed CAZymes in the rumen were GH5, GH9, GH45 and GH48, primarily from Ruminococcus and Fibrobacter (Dai et al., Reference D’Amore, Ijaz, Schirmer, Kenny, Gregory, Darby, Shakya, Podar, Quince and Hall2015). The metagenomic study of rumen contents from dairy cows fed a high-forage diet by Hess and colleagues likewise found that the GH5 and GH9 CAZyme families putatively involved in cellulose digestion were abundant, whereas GH45 and GH48 were not (Hess et al., Reference Hess, Sczyrba, Egan, Kim, Chokhawala, Schroth, Luo, Clark, Chen, Zhang, Mackie, Pennacchio, Tringe, Visel, Woyke, Wang and Rubin2011). Differences in gene abundances observed between DNA- or RNA-based studies have been reinforced by several other rumen metatranscriptome studies, thus highlighting the importance of combined approaches (Qi et al., Reference Qi, Wang, O’Toole, Barboza, Ungerfeld, Leigh, Selinger, Butler, Tsang and McAllister2011; Shinkai et al., Reference Shinkai, Mitsumori, Sofyan, Kanamori, Sasaki, Katayose and Takenaka2016; Comtet-Marre et al., Reference Comtet-Marre, Parisot, Lepercq, Chaucheyras-Durand, Mosoni, Peyretaillade, Bayat, Shingfield, Peyret and Forano2017). These studies have pointed to the rumen fungi, Prevotella sp., Ruminococcus sp. and Fibrobacter sp. as being responsible for the production of a large proportion of the rumen CAZymes involved in lignocellulose digestion (Dai et al., Reference D’Amore, Ijaz, Schirmer, Kenny, Gregory, Darby, Shakya, Podar, Quince and Hall2015; Shinkai et al., Reference Shinkai, Mitsumori, Sofyan, Kanamori, Sasaki, Katayose and Takenaka2016; Comtet-Marre et al., Reference Comtet-Marre, Parisot, Lepercq, Chaucheyras-Durand, Mosoni, Peyretaillade, Bayat, Shingfield, Peyret and Forano2017).
Metagenomics and metatranscriptomics have also been used to examine the link between feed efficiency (based on residual feed intake (RFI)) and the rumen microbiome (Shabat et al., Reference Shabat, Sasson, Doron-Faigenboim, Durman, Yaacoby, Berg Miller, White, Shterzer and Mizrahi2016; Li and Guan, Reference Li and Guan2017). With both approaches it was found that cattle with low feed efficiency have a greater diversity of metabolic pathways than those that are more efficient (Li and Guan, Reference Li and Guan2017). Increasing the diversity of metabolic pathways present in the rumen seems to increase the likelihood of carbon being shuttled into pathways that are less energy efficient, reducing the amount of energy the host can derive from metabolism. In contrast, highly efficient animals seem to have microbial communities that direct more carbon into fewer, more efficient metabolic pathways which lower the production of less metabolically valuable end products, such as methane (Shabat et al., Reference Shabat, Sasson, Doron-Faigenboim, Durman, Yaacoby, Berg Miller, White, Shterzer and Mizrahi2016; Li and Guan, Reference Li and Guan2017). Both of these studies included a total mixed ration (TMR) containing significant levels of concentrate so it would be interesting to examine whether this relationship holds in cattle fed a diet with high forage and little to no concentrate. We recently conducted a study with cattle fed a 70% barley straw diet and observed apparent neutral detergent fibre digestibility ranging from 42.2% to 61.1% (Ribeiro et al., Reference Ribeiro, Oss, He, Gruninger, Elekwachi, Forster, Yang, Beauchemin and McAllister2017). Fibre digestibility in this study was highly correlated with Metadinium protozoa. This suggests that there is high variability in the ability of individual cattle to digest recalcitrant fibre, and that more work should be done towards defining the role of protozoa and fungi in this process. The studies conducted to date suggest that lower microbial diversity in the rumen is linked to higher feed efficiency. It is not known how this might influence other important aspects of the animal, including adaptation to diet changes, resilience to illness or recovery from acidosis, and further research into examining the link between feed efficiency, the rumen microbiome, and the host is warranted.
Applying a combination of metagenomics and metatranscriptomics has enabled researchers to link members of the rumen microbiome in sheep that differ in methane production levels to rumen VFA profiles in these animals (Franzosa et al., Reference Franzosa, Morgan, Segata, Waldron, Reyes, Earl, Giannoukos, Boylan, Ciulla, Gevers, Izard, Garrett, Chan and Huttenhower2014; Kamke et al., Reference Kamke, Kittelmann, Soni, Li, Tavendale, Ganesh, Janssen, Shi, Froula, Rubin and Attwood2016). In another study, a systems biology approach using 16S rRNA amplicon sequencing, metaproteomics and metabolomics found that the starch content of different forage-based diets accounted for an increase in the abundance and metabolic activity of bacteria in the family Succinivibrionaceae, particularly starch-utilizing Succinionas amylolytica and Ruminobacter amylophilus (Deusch et al., Reference Dias, Marcondes, Noronha, Resende, Machado, Mantovani, Dill-McFarland and Suen2017). Further efforts are required in this area to link shifts in microbiome composition and biological activity to host phenotype and host physiology. Another area requiring further efforts to better understand is how the rumen microbiome changes over time. Many -omics studies of the rumen to date have only examined samples taken during a limited time scale. There is a need for longitudinal studies to examine how the microbiome changes over time as temporal changes also influence the development of the microbiome and ultimately its impact on host productivity (Shaani et al., Reference Shaani, Zehavi, Eyal, Miron and Mizrahi2018).
Technical challenges of sequence-based studies of the rumen
It is well known that sequencing different variable regions of the 16S rRNA gene influences the taxonomy assigned to those sequences; this is one of the most common sources of variation between microbiome studies (Pollock et al., Reference Pollock, Glendinning, Wisedchanwet and Watson2018). Unfortunately, there is still no clear consensus as to which variable region of the most commonly used amplicon, the 16S rRNA gene, provides the greatest classification accuracy (Claesson et al., Reference Claesson, Wang, O’sullivan, Greene-Diniz, Cole, Ross and OTtoole2010; D’Amore et al., Reference Dai, Tian, Li, Su, Wang, Zhao, Liu, Luo, Liu and Zheng2016; Yang et al., Reference Yang, Wang and Qian2016). The most commonly used region for Illumina-based sequencing is the V4 region (Caporaso et al., Reference Caporaso, Lauber, Walters, Berg-Lyons, Huntley, Fierer, Owens, Betley, Fraser, Bauer, Gormley, Gilbert, Smith and Knight2012). Sequencing the 16S rRNA V4 region has consistently shown the highest similarity to expected taxonomic distribution, and gives results similar to those obtained from shotgun metagenomics, a sequencing approach not subject to PCR bias (Caporaso et al., Reference Caporaso, Lauber, Walters, Berg-Lyons, Huntley, Fierer, Owens, Betley, Fraser, Bauer, Gormley, Gilbert, Smith and Knight2012; Tremblay et al., Reference Tremblay, Singh, Fern, Kirton, He, Woyke, Lee, Chen, Dangl and Tringe2015; D’Amore et al., Reference Dai, Tian, Li, Su, Wang, Zhao, Liu, Luo, Liu and Zheng2016).
The recent focus on using short-read technologies to characterize microbial communities has resulted in less full-length 16S rRNA sequences. Full-length sequences improve the depth of phylogenetic analyses and are useful for the design of lineage-specific PCR primers and fluorescent in situ hybridization (FISH) probes (Schloss et al., Reference Schloss, Jenior, Koumpouras, Westcott and Highlander2016). To address this limitation, PacBio SMRT long-read technology has been used to generate full-length 16S rRNA sequences with error rates of 0.027% to –0.69%: rates comparable to Illumina technology. Shorter reads from Illumina sequencing can characterize microbial communities at the OTU level but are less accurate at characterizing communities at the genus and species level (Liu et al., Reference Liu, Lozupone, Hamady, Bushman and Knight2007; Soergel et al., Reference Soergel, Dey, Knight and Brenner2012). Myer et al. (Reference Myer, Kim, Freetly and Smith2016) compared whether full-length 16S rRNA reads (V1–V8) sequenced using PacBio were able to classify rumen community members at greater depth than shorter V1–V3 reads sequenced using Illumina Mi-Seq. They found that while the two platforms revealed similar microbial OTUs, species richness, Good’s coverage and Shannon diversity metrics, the Pac-Bio improved the taxonomic depth. Adoption of Oxford Nanopore MinION will undoubtedly accelerate the use of full-length sequencing in the generation of metagenomic assemblies, as it has already shown promise as a tool to assemble whole chromosomes from complex rumen metagenomes (Stewart et al., Reference Stewart, Auffret, Warr, Wiser, Walker and Watson2018b).
Several factors can generate variation between metagenomics studies, including sampling method, sampling site, number of samples, library preparation (Clooney et al., Reference Clooney, Fouhy, Sleator, O’Driscoll, Stanton, Cotter and Claesson2016), sequencing technology (D’Amore et al., Reference Dai, Tian, Li, Su, Wang, Zhao, Liu, Luo, Liu and Zheng2016), differences in bioinformatic pipelines (Pollock et al., Reference Pollock, Glendinning, Wisedchanwet and Watson2018), differences in databases (Soergel et al., Reference Soergel, Dey, Knight and Brenner2012; Schloss et al., Reference Schloss, Jenior, Koumpouras, Westcott and Highlander2016) and even sequencing facility (Kim and Yu, Reference Kim and Yu2014). It is also well known that the microbiome among individuals varies even when other factors such as diet composition are kept constant (Flores et al., Reference Flores, Caporaso, Henley, Rideout, Domogala, Chase, Leff, Vazquez-Baeza, Gonzalez, Knight, Dunn and Fierer2014). Therefore, researchers should keep in mind that comparisons of relative abundances of OTUs and taxa between different studies can, in principle, lead to erroneous conclusions. This is particularly the case for OTUs or taxa that account for a relatively low percentage of the bacterial population (Kim and Yu, Reference Kim and Yu2014).
Development of the rumen microbiome in young ruminants
A number of studies have examined the development of the rumen microbiome from neonate to the mature animal (Jami et al., Reference Jami, Israel, Kotser and Mizrahi2013; Jiao et al., Reference Jiao, Lu, Forster, Zhou, Wang, Kang and Tan2016; Meale et al., Reference Meale, Li, Azevedo, Derakhshani, Plaizier, Khafipour and Steele2016; Wang et al., Reference Wang, Zhao, Wang, Zhang, Zheng and Wang2016a). Shortly after birth, there is a shift in the bacterial community from a highly variable aerobic and facultative anaerobic community to a diverse community of mainly obligate anaerobes (Jami et al., Reference Jami, Israel, Kotser and Mizrahi2013). This transition climaxes at approximately 3 months of age and is characterized by an increase in the abundance of Bacteroidetes and a decrease in the abundance of Proteobacteria (Jami et al., Reference Jami, Israel, Kotser and Mizrahi2013; Jiao et al., Reference Jiao, Lu, Forster, Zhou, Wang, Kang and Tan2016; Wang et al., Reference Wang, Zhao, Wang, Zhang, Zheng and Wang2016a). Within the phylum Firmicutes, Bacteroides is the most abundant genus in newborn calves, whereas in 2-month-old calves, Prevotella is the principal genera observed (Jami et al., Reference Jami, Israel, Kotser and Mizrahi2013). This microbial succession was linked to alterations in the diet associated with a shift from a primarily milk-based diet to a post-weaning forage-based diet (Jami et al., Reference Jami, Israel, Kotser and Mizrahi2013; Meale et al., Reference Meale, Li, Azevedo, Derakhshani, Plaizier, Khafipour and Steele2016; Wang et al., Reference Wang, Zhao, Wang, Zhang, Zheng and Wang2016a). Undoubtedly, changes in diet and feeding behaviour also contribute to changes in rumen morphology and function and ultimately the rumen microbiome. For example, in nursing ruminants, a specialized structure called the reticular groove directs milk to flow into the abomasum, by-passing the rumen. Interestingly, the celluloytic bacteria Ruminococcus flavefaciens and R. albus were detected in the rumen of 1- and 3-day-old calves respectively, while Fibrobacter succinogenes was not detectable in the rumen until after 2 months of age (Jami et al., Reference Jami, Israel, Kotser and Mizrahi2013). Archaeal, bacterial and fungal communities can be detected after 7 days in the rumen of calves that have only received milk, with populations varying thereafter with both age of the calf and the type of diet (Dias et al., Reference de Menezes, Lewis, O’Donovan, O’Neill, Clipson and Doyle2017).
Impact of forage on the rumen microbiome
After weaning, the diet of the ruminant shifts to one primarily consisting of high-fibre forages and grains. The ability of ruminants to digest high-fibre forages is due to the enzymatic activity of the rumen microbiome. Alterations in the chemical composition of forage, the addition of concentrates or the addition of other additives can alter the composition of the rumen microbiome. One study examining the impact that differences in the composition of forage have on the rumen microbiome of dairy cows found that animals fed pasture or TMR diets did not have obvious differences at the phylum level, with Firmicutes and Bacteroidetes representing approximately 80% of the total rumen microbiome sequences (de Menezes et al., Reference Deusch, Camarinha-Silva, Conrad, Beifuss, Rodehutscord and Seifert2011). This did not hold true at higher levels of taxonomic resolution as there were clear differences between pasture and TMR diets for Bacteroidetes, Firmicutes, Fibrobacteres and Proteobacteria (de Menezes et al., Reference Deusch, Camarinha-Silva, Conrad, Beifuss, Rodehutscord and Seifert2011). Interestingly, the family Fibrobacteriaceae accounted for almost 10% of the sequences in solid rumen contents from cattle fed TMR diets compared to ~3% in cattle on pasture. The observed increase in Fibrobacteriaceae was suggested to be due to an increased level of lignocellulose in the diet from the addition of straw (3% of diet DM) to the diet. This hypothesis is supported by Ribeiro et al. (Reference Ribeiro, Oss, He, Gruninger, Elekwachi, Forster, Yang, Beauchemin and McAllister2017) who found that Fibrobacteraceae accounted for 25% of the OTUs in rumen solids from heifers fed a 70% barley straw diet. In other studies, Fibrobacteriaceae were only a minor component in cattle fed TMR diets indicating that variability between animals likely exists (Callaway et al., Reference Callaway, Dowd, Edrington, Anderson, Krueger, Bauer, Kononoff and Nisbet2010; Zened et al., Reference Zened, Combes, Cauquil, Mariette, Klopp, Bouchez, Troegeler-Meynadier and Enjalbert2013).
Looking specifically at the effect of forage source in the diet, Kong et al. (Reference Kong, Teather and Forster2010) observed that cows fed an alfalfa hay-based diet had different bacterial communities and greater species richness than cows fed a triticale straw-based diet. This seems to be related to the greater nutrient availability in alfalfa hay for ruminal microbial populations, as compared to the restricted nutrient content of triticale straw. Prevotella were the most abundant genera (comprising 14% to 22% of the total clones) in both diets, suggesting that this genus plays a fundamental role in the rumen ecosystem (Kong et al., Reference Kong, Teather and Forster2010). Changing the forage source (e.g. corn silage, grass silage or grass hay) in a 48% forage:52% concentrate diet also altered the ruminal bacterial population of Jersey cows (Deusch et al., Reference Dias, Marcondes, Noronha, Resende, Machado, Mantovani, Dill-McFarland and Suen2017). A higher abundance of Proteobacteria and Succinivibrionaceae were observed in the rumen of cows fed corn silage, consistent with the higher content of non-fibre carbohydrates in this diet as compared to grass diets. In agreement with previous studies, Prevotellaceae was the most abundant family detected (Deusch et al., Reference Dias, Marcondes, Noronha, Resende, Machado, Mantovani, Dill-McFarland and Suen2017). Seasonal changes in pasture quality throughout the year also promoted changes in the ruminal bacterial communities attached to solid digesta collected from dairy cows grazing rye-grass/clover pasture (Noel et al., Reference Noel, Attwood, Rakonjac, Moon, Waghorn and Janssen2017). Overall, the most abundant bacterial groups were uncharacterized genera in the order Clostridiales accounting for 22.9% of OTUs, followed by uncharacterized genera in the family Lachnospiraceae (12.2%) and Butyrivibrio (10.2%), Ruminococcus (7.6%) and Prevotella (6.7%). This study showed that the rumen bacterial community can adapt to relatively small changes in forage quality, and to changes in the forage:grain ratio. Although small seasonal changes in ruminal bacterial communities were observed, principal members of the community remained consistent, suggesting their central role in feed degradation regardless of changes in pasture quality (Noel et al., Reference Noel, Attwood, Rakonjac, Moon, Waghorn and Janssen2017).
The Prevotellaceae are one of the most abundant microbes in the rumen and are known to have an extensive metabolic repertoire that enables the utilization of a wide range of substrates. The cellulolytic rumen bacteria Ruminococcus flavefaciens, Ruminococcus albus and Fibrobacter succinogenes are most abundant when the diet is mainly forage, but changes in the type of forage in the diet do not seem to alter the relative abundance of these bacteria (Deusch et al., Reference Dias, Marcondes, Noronha, Resende, Machado, Mantovani, Dill-McFarland and Suen2017). It has been suggested that these fibrolytic bacteria may be regarded as ’keystone’ species and that their numbers alone do not clearly represent their contribution to rumen fibre digestion (Ze et al., Reference Ze, Le Mougen, Duncan, Louis and Flint2013; Creevey et al., Reference Creevey, Kelly, Henderson and Leahy2014). Recent metatranscriptomic analysis of the rumen contents of cattle fed a 50:50 forage:concentrate TMR supports the pivotal role of these well-characterized fibrolytic bacteria (Prevotella, Ruminoccocus and Fibrobacter) in fibre degradation (Comtet-Marre et al., Reference Comtet-Marre, Parisot, Lepercq, Chaucheyras-Durand, Mosoni, Peyretaillade, Bayat, Shingfield, Peyret and Forano2017). Several studies have also found low levels of these well-known cellulolytic bacteria in cattle fed a high-forage diet (Creevey et al., Reference Creevey, Kelly, Henderson and Leahy2014; Tapio et al., Reference Tapio, Fischer, Blasco, Tapio, Wallace, Bayat, Ventto, Kahala, Negussie and Shingfield2017). Interestingly, Tapio et al. (Reference Tapio, Fischer, Blasco, Tapio, Wallace, Bayat, Ventto, Kahala, Negussie and Shingfield2017) observed that increased forage content resulted in an increase the abundance of many uncharacterized bacteria within the Clostridiaceae and Lachnospiraceae families. Similar results were also observed by Henderson et al. (Reference Henderson, Cox, Ganesh, Jonker, Young and Janssen2015) and Noel et al. (Reference Noel, Attwood, Rakonjac, Moon, Waghorn and Janssen2017), which suggests that there are largely undescribed bacteria in the rumen that may have an important role in fibre degradation. Further research characterizing these unknown bacteria using approaches that include ultra deep sequencing, the use of single-cell genomics or increased efforts at culturing rumen microbes is needed to obtain a more complete understanding of fibre digestion in the rumen (Hosokawa et al., Reference Hosokawa, Nishikawa, Kogawa and Takeyama2017; Huws et al., Reference Huws, Mayorga, Theodorou, Kim, Cookson, Newbold and Kingston-Smith2018).
Colonization of feed in the rumen
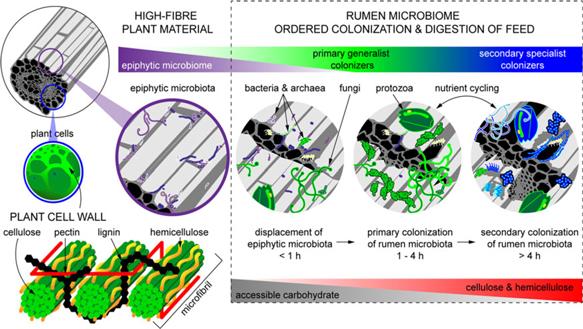
When feed enters the rumen, it is rapidly colonized by rumen microbes, and the digestion of the plant cell wall begins within minutes (Edwards et al., Reference Edwards, Huws, Kim and Kingston-Smith2007; Edwards et al., Reference Edwards, Kingston-Smith, Jimenez, Huws, Skot, Griffith, McEwan and Theodorou2008). Bacteria preferentially attach to damaged sites on the plant surface, whereas fungi are able to physically disrupt the ingested material via rhizoidal growth. Microbial attachment is absolutely essential for the development of the complex microbial populations required for feed digestion in the rumen and occurs via a multistep process: (1) displacement of the epiphytic microbiome by rumen microbes (time <1 h), (2) establishment of a primary colonizing community of generalist microbes that primarily metabolize accessible carbohydrates (time 1 h to 4 h), (3) loss of some primary colonizers and selection of secondary colonizers specialized in digesting hemicellulose and cellulose (time > 4 h; Figure 2) (Piao et al., Reference Piao, Lachman, Malfatti, Sczyrba, Knierim, Auer, Tringe, Mackie, Yeoman and Hess2014; Huws et al., Reference Huws, Edwards, Creevey, Rees Stevens, Lin, Girdwood, Pachebat and Kingston-Smith2016; Mayorga et al., Reference Mayorga, Kingston-Smith, Kim, Allison, Wilkinson, Hegarty, Theodorou, Newbold and Huws2016). Within this community are taxa including Butyrivibrio, Fibrobacter, Olsenella and Prevotella that do not change significantly in abundance during primary and secondary colonization (Huws et al., Reference Huws, Edwards, Creevey, Rees Stevens, Lin, Girdwood, Pachebat and Kingston-Smith2016; Liu et al., Reference Liu, Zhang, Xue, Zhu and Mao2016). Populations of Prevotella peak on the surface of fibre within 1 h in the rumen (Huws et al., Reference Huws, Edwards, Creevey, Rees Stevens, Lin, Girdwood, Pachebat and Kingston-Smith2016). Prevotella sp. have been noted to be ubiquitous in the rumen environment and have a wide range of metabolic capabilities (Petri et al., Reference Petri, Schwaiger, Penner, Beauchemin, Forster, McKinnon and McAllister2013; Rubino et al., Reference Rubino, Carberry, Waters, Kenny, McCabe and Creevey2017). This metabolic flexibility likely explains the involvement of Prevotella in both primary and secondary colonization of feed as it can utilize soluble carbohydrates, pectins, proteins and hemicellulose (Huws et al., Reference Huws, Edwards, Creevey, Rees Stevens, Lin, Girdwood, Pachebat and Kingston-Smith2016). Populations of Clostridia (Pseudobutyrivibrio, Roseburia and Ruminococcus sp.) peak after Prevotella, perhaps due to their role in targeting cellulose (Piao et al., Reference Piao, Lachman, Malfatti, Sczyrba, Knierim, Auer, Tringe, Mackie, Yeoman and Hess2014; Rubino et al., Reference Rubino, Carberry, Waters, Kenny, McCabe and Creevey2017). In accordance with this model, it has been observed that little biomass is degraded during primary colonization, with the majority of degradation occurring after this (Piao et al., Reference Piao, Lachman, Malfatti, Sczyrba, Knierim, Auer, Tringe, Mackie, Yeoman and Hess2014; Huws et al., Reference Huws, Edwards, Creevey, Rees Stevens, Lin, Girdwood, Pachebat and Kingston-Smith2016). It has also been hypothesized that decreasing microbial richness during the shift from primary to secondary colonization is due to niche specialization of Clostridiales and Bacteroidales, which play a specific role in lignocellulose degradation during secondary colonization (Huws et al., Reference Huws, Edwards, Creevey, Rees Stevens, Lin, Girdwood, Pachebat and Kingston-Smith2016; Mayorga et al., Reference Mayorga, Kingston-Smith, Kim, Allison, Wilkinson, Hegarty, Theodorou, Newbold and Huws2016; Rubino et al., Reference Rubino, Carberry, Waters, Kenny, McCabe and Creevey2017).
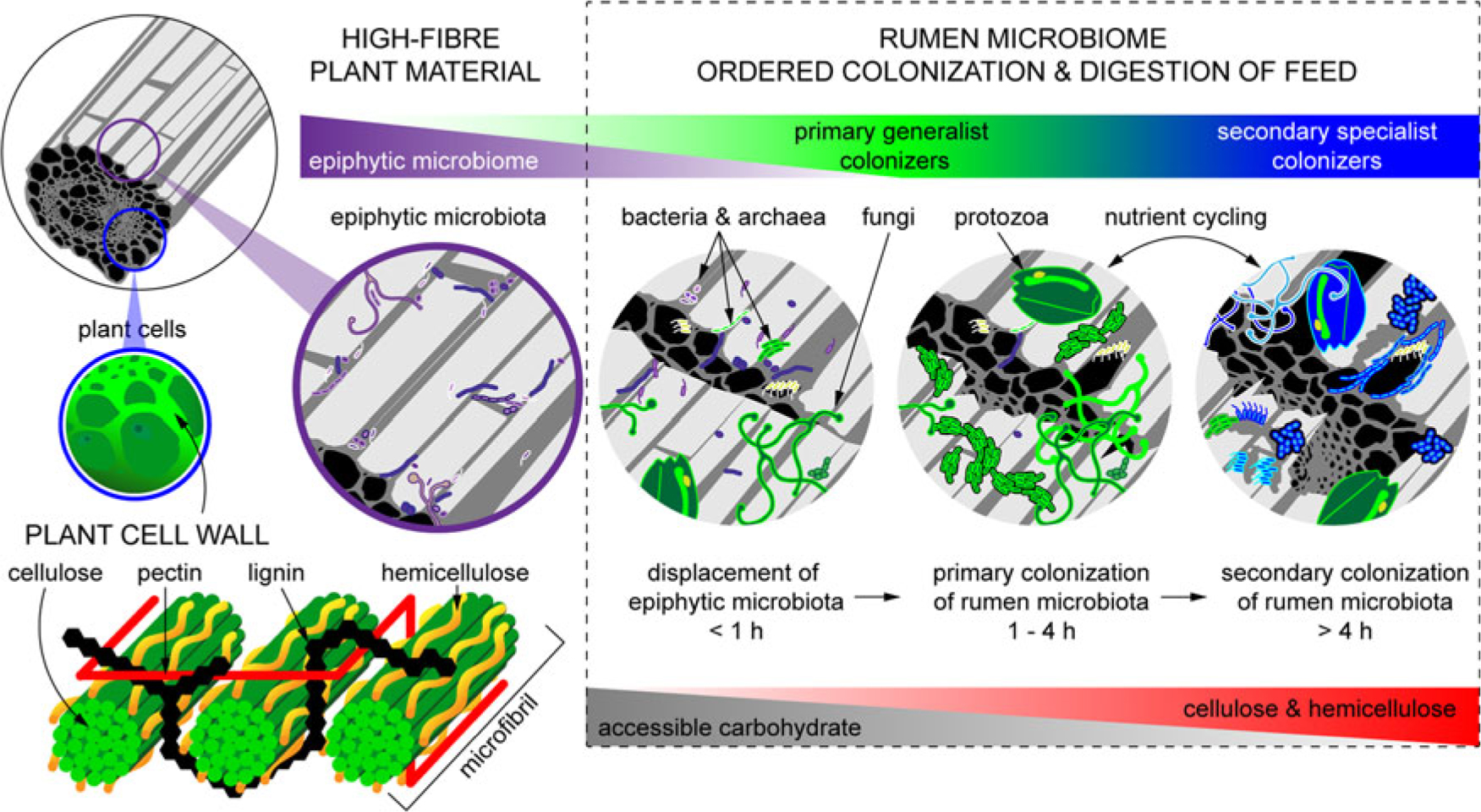
Figure 2 Fibrous material in plants is colonized by a natural epiphytic microbiome which colonizes the forage while it grows in the field. This microbiome can be altered if the plant is ensiled prior to consumption. Upon consumption this epiphytic population is displaced by primary colonizing bacteria that ferment primarily sugars and soluble proteins. These primary colonizers are in turn replaced by secondary colonizers which play a more active role in the digestion of structural carbohydrates in plant cell walls.
The structure and chemical composition of feedstuffs is a key determinant defining which microbes participate in the ordered colonization of feed (Huws et al., Reference Huws, Creevey, Oyama, Mizrahi, Denman, Popova, Muñoz-Tamayo, Forano, Waters, Hess, Tapio, Smidt, Krizsan, Yáñez-Ruiz, Belanche, Guan, Gruninger, McAllister, Newbold, Roehe, Dewhurst, Snelling, Watson, Suen, Hart, Kingston-Smith, Scollan, do Prado, Pilau, Mantovani, Attwood, Edwards, McEwan, Morrisson, Mayorga, Elliott and Morgavi2014; Liu et al., Reference Liu, Zhang, Xue, Zhu and Mao2016). The shifting nature of the community involved in feed digestion underlies the need to allow animals to adapt to shifts in diet, particularly from forage-based to concentrate-based diets. This transition has been reported to require approximately 14 days and involves significant changes in ruminal microbial populations (Petri et al., Reference Petri, Schwaiger, Penner, Beauchemin, Forster, McKinnon and McAllister2013; Pitta et al., Reference Pitta, Pinchak, Indugu, Vecchiarelli, Sinha and Fulford2014; Kittelmann et al., Reference Kittelmann, Kirk, Jonker, McCulloch and Janssen2015). Transition from a mainly forage diet to a high-concentrate diet has been shown in several studies to lead to an increase in the abundance of Bacteroidetes and a decrease in Firmicutes and Proteobacteria (Petri et al., Reference Petri, Schwaiger, Penner, Beauchemin, Forster, McKinnon and McAllister2013; Kittelmann et al., Reference Kittelmann, Kirk, Jonker, McCulloch and Janssen2015). Inclusion of cereal grain in the diet results in higher levels of ruminal starch, which promotes the growth of amylolytic bacteria such as Ruminobacter sp. and Succinivibrio sp. (Kittelmann et al., Reference Kittelmann, Kirk, Jonker, McCulloch and Janssen2015). In forage diets, there is a significant increase in fibrolytic microbes, including anaerobic fungi, Ruminococcaceae, as well as non-fibrolytic bacteria such as Succiniclasticum sp. (Kumar et al., Reference Kumar, Indugu, Vecchiarelli and Pitta2015). Anaerobic fungi are highly responsive to increases in concentrate levels, and their abundance decreases with increasing starch in the diet (Belanche et al., Reference Belanche, de la Fuente and Newbold2012). However, alterations in diet seem to have less impact on the abundance of methanogenic archaea (Kumar et al., Reference Kumar, Indugu, Vecchiarelli and Pitta2015), possibly because of the more uniform availability of reducing equivalents in the rumen and their ability to avoid washout through physical associations such as those observed with protozoa (Levy and Jami, Reference Ley, Lozupone, Hamady, Knight and Gordon2018).
Role of diet in dysbioses of the rumen microbiome
Rapid diet changes that do not enable the rumen microbes to adapt can lead to digestive upset and other health problems. For example, frothy bloat occurs in ruminants consuming a diet with high levels of rapidly digestible soluble protein and carbohydrate (Grilli et al., Reference Grilli, Mrázek, Fliegerová, Kopečný, Lama, Cucchi, Sosa and Arenas2016). Bloat can be caused by abrupt shifts to protein-rich leguminous forages, as well as by rapid shifts to extremely high levels of concentrate. The inability of the animal to release gas produced by rumen fermentation due to impairment of eructation can prevent the contraction of the diaphragm and, if not relieved, lead to suffocation. Bloat in cattle grazing wheat pastures was linked to increased production of biofilm-associated mucopolysaccharide in the rumen resulting from diet-induced shifts in the rumen bacterial population (Min et al., Reference Min, Pinchak, Anderson, Fulford and Puchala2006). Frothy bloat in goats resulted in long-lasting changes in the structure of the rumen microbial community, which persisted even after the clinical manifestations of bloat ceased (Grilli et al., Reference Grilli, Mrázek, Fliegerová, Kopečný, Lama, Cucchi, Sosa and Arenas2016). A metagenomic study examining the microbial role in frothy wheat bloat in cattle revealed disruption in symbiotic relationships among microbial taxa, increased abundance of methanogenic archaea and a reduction in the abundance and diversity of CAZymes, suggesting an alteration in carbohydrate metabolism (Pitta et al., Reference Pitta, Pinchak, Dowd, Dorton, Yoon, Min, Fulford, Wickersham and Malinowski2016).
Clinical (rumen pH < 5.2) and sub-clinical acidosis (rumen pH 5.2 to 5.6 for at least 3 h) are conditions that can also be linked with shifts in the rumen microbiome (Owens et al., Reference Owens, Secrist, Hill and Gill1998). The classical view of acidosis is that rapid fermentation of feed results in the production of VFAs at a rate greater than they can be absorbed across the rumen wall, or pass through the rumen to the lower digestive tract. As a consequence, the pH of the rumen decreases to a point where cellulolytic bacteria are inhibited and acid-tolerant, lactate-producing bacteria, particularly Streptococcus bovis and Lactobacillus sp., predominate (Khafipour et al., Reference Khafipour, Li, Plaizier and Krause2009). These conditions are correlated with a reduction in species richness and diversity of the rumen microbiota (Khafipour et al., Reference Khafipour, Li, Plaizier and Krause2009; Petri et al., Reference Petri, Schwaiger, Penner, Beauchemin, Forster, McKinnon and McAllister2013; McCann et al., Reference McCann, Luan, Cardoso, Derakhshani, Khafipour and Loor2016; Plaizier et al., Reference Plaizier, Li, Tun and Khafipour2017). This can be modulated, however, by the activity of the lactic acid–utilizing bacteria Megasphaera elsdenii and Selenomonas ruminantium. The activity of these bacteria is increased at high lactate concentrations, and consequently, lactic acid concentrations often do not reach levels associated with clinical acidosis (McCann et al., Reference McCann, Luan, Cardoso, Derakhshani, Khafipour and Loor2016). Acidosis is also associated with higher levels of Escherichia coli in the rumen and production of ruminal lipopolysaccharides (LPS) which contributes to systemic inflammation in grain-fed cattle suffering from clinical acidosis (Plaizier et al., Reference Plaizier, Li, Danscher, Derakshani, Andersen and Khafipour2016). Indeed, profiling of the E. coli population in acidotic rumen contents revealed a shift in the population towards E. coli isolates with unique virulence factors that trigger inflammatory responses (Khafipour et al., Reference Khafipour, Plaizier, Aikman and Krause2011). Despite acidosis-associated changes in the rumen microbiome, the core microbial community remains seemingly unaltered, and recovery of the rumen microbiome has been shown to occur approximately 1 week following clinical or subclinical acidosis (Petri et al., Reference Petri, Schwaiger, Penner, Beauchemin, Forster, McKinnon and McAllister2013).
Future opportunities
Although great progress has been made in the past decade towards a better understanding of the rumen microbiome under a range of conditions, there is still a great deal that is not understood. Efforts must be made to continue to culture rumen bacteria that have to date resisted cultivation. The integration of genomic information from efforts such as Hungate1000 will hopefully enable researchers to design novel growth media with essential co-factors and energy sources that these uncultured organisms require. Although researchers have attempted to manipulate the rumen microbiome, the resiliency of the community has limited the success of such efforts (Weimer, Reference Wenner, Wagner and Firkins2015). We have recently attempted to manipulate the rumen of cattle by repeated inoculation with bison rumen contents (Ribeiro et al., Reference Ribeiro, Oss, He, Gruninger, Elekwachi, Forster, Yang, Beauchemin and McAllister2017). Although initial changes in microbial population were observed, we were unsuccessful in improving fibre degradation. At present, it is unclear whether there is a point in the animal’s life that the rumen microbiome becomes irreversibly programmed. Efforts towards a better understanding of the development of the rumen microbial community in young ruminants have provided some insights, but more needs to be done to determine if probiotics or other approaches can be used to generate a more resilient rumen microbial community that enhances the efficiency and health of the host.
Interactions between the host and the rumen microbiome are not well understood. Future studies need to not only consider interactions among microbes but also how microbial metabolites alter host gene expression in cells and tissues such as the rumen epithelium, liver and immune system. Recent work examining the connection between the immune system in ruminants has found host-specific interaction between salivary immunoglobulin IgA (Fouhse et al., Reference Fouhse, Smiegielski, Tuplin, Guan and Willing2017) and upregulation of several ruminal epithelial Toll-like receptors (TLRs) in response to high-grain diets (Liu et al., Reference Liu, Bian, Zhu and Mao2015). TLRs are known to be involved in recruitment of immune cells and the production of inflammatory cytokines. This hints at how changes in the rumen environment can cause systemic changes in the host and vice versa.
Conclusion
Over the past decade, significant research has been directed towards understanding both the composition of the rumen microbiome and how it affects the growth and health of the host. It is clear that diet can have dramatic effects on the taxonomic composition of the rumen, and that these changes are linked to the nature of the nutrients in the rumen. More efforts are needed to incorporate information on the nature of the microbes present and their metabolic activity through complementary -omics approaches. Efforts must now be made to understand how this community interacts with the physiological function of the rumen and the host, ultimately influencing both the health and productivity of the animal.
Acknowledgements
The authors wish to acknowledge the Beef Cattle Research Council of the Growing Forward II program of Agriculture and Agri-Food Canada for support of their work. We also wish to thank Edith Vale for generating the confocal microscopy pictures of rumen protozoa.
Declaration of interest
The authors have no conflicts to declare.
Ethics statement
This review did not require ethics approval.
Software and data repository resources
None of the data were deposited in an official repository.