

The Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study (Reference Lieberman, Stroup and McEvoyLieberman 2005) is arguably the highest-profile psychopharmacology trial ever, for this was a study with a sting in its message – that, in effect, for over a decade, psychiatry had got it wrong (Reference OwensOwens 2008). There are still those who reject such radical interpretation (Reference Naber and LambertNaber 2009) but much of CATIE’s prodigious output has been aimed at addressing potential design flaws and, overall, it has stood up well to a level of scrutiny most studies avoid. Furthermore, it is now only one of a series of pragmatic studies of differing designs and outcomes, and not sponsored by industry (Reference Jones, Barnes and DavisJones 2006; Reference Kahn, Fleischhacker and BoterKahn 2008; Reference Fischer-Barnicol, Lanquillon and HaenFischer-Barnicol 2008; Reference Sikich, Frazier and McClellanSikich 2008), pointing to the unavoidable conclusion that the distinction between ‘typical’ and ‘atypical’ antipsychotics is artificial. This has led some to argue that any term implying a dichotomous split in antipsychotic drugs, such as ‘atypical’ or ‘second-generation’, should be banned from teaching and practice (Reference Fischer-Barnicol, Lanquillon and HaenFischer-Barnicol 2008; Reference OwensOwens 2008; Reference Leucht, Kissling and DavisLeucht 2009), allowing the entire field of antipsychotics, old and new, to become available for equal consideration in individualised treatment planning.
This overlooks an obvious fact – that a generation of trainees has now reached seniority in psychiatry without gaining much, if any, experience in the use of antipsychotics available before the mid-1990s. This is important, for one lesson from CATIE might be that we were not very good at using the older drugs in the first place. This article offers a brief clinical overview of the so-called ‘first-generation’ (‘conventional’ or ‘typical’) antipsychotics currently available in the UK (Box 1). While a ‘clinical pharmacology’ approach would be ideal, formal study of these compounds was generally sparse, so a large part of what follows encompasses my own experience as a clinician now sufficiently ‘senior’ to have accrued some years of their utilisation.
BOX 1 Older (‘first-generation’) antipsychotics currently available in the UK (oral formulationsFootnote a)
Phenothiazines
-
• Chlorpromazine
-
• PromazineFootnote b
-
• Levomepromazine (methotrimeprazine)
-
• Pericyazine (periciazine)
-
• Perphenazine
-
• ProchlorperazineFootnote b
-
• Trifluoperazine
Thioxanthenes
-
• Flupentixol (flupenthixol)
-
• Zuclopenthixol (zuclopentixol)Footnote c
Butyrophenones and diphenylbutylpiperidines
-
• Haloperidol
-
• BenperidolFootnote b
-
• Pimozide
Substituted benzamides
-
• Sulpiride
a The following are also available as depots: flupentixol, zuclopenthixol (esterified with decanoic acid), fluphenazine, pipotiazine, haloperidol.
b Uncommonly used as antipsychotics or with limited/no data for efficacy.
c Also esterified with acetic acid.
The article focuses on oral preparations, as these still comprise the ‘essential’ first step in the treatment of most psychotic illnesses and are the group to have suffered most neglect with the introduction of new products.
History
The surgeon Henri Laborit was given belated access to the newly developed ‘chloropromazine’ (sic) in 1951, after psychiatrists had noticed only sedation (Reference OwensOwens 1999). He detected something different – ‘detachment’, ‘indifference’, an affective change that was unique and distinct from sedation. And he noted this (subsequently termed ataraxy) after single and, by our standards, small doses (50–75 mg usually administered by intramuscular injection). ‘Ataraxics’ lost out to ‘neuroleptics’ in christening the new class of drugs, which was unfortunate, for ataraxy refers to a particular mental state effect (possibly the target action) that is key to understanding the clinical pharmacology of the entire class, whereas neuroleptic points to a group of side-effects. The loss of the concept is the more regrettable for there has been hardly any investigation of the neural basis of ataraxy, an astonishing oversight of basic pharmacological mechanisms in such a widely utilised class of drugs. By the late 1950s, the subjective features comprising ataraxy had become subsumed under mild/early Parkinsonism, an assumption as significant if it is wrong as if it is correct.
The importance of this lies in the realisation that antipsychotics (as the neuroleptics came to be called) may create, as class effects, subjective states that, first, are difficult if not impossible to distinguish from illness-related disorders and second, can have detrimental consequences for quality of life and adherence. Antipsychotics can be difficult drugs for patients to take but can also be difficult for doctors to prescribe sensitively.
A note on ‘indications’
In recent years, the pharmaceutical industry has moved to extend indications for new anti-psychotics, especially to certain mood states. This has highlighted doctors’ confusion about the nature of drug licensing.
Licensing is not about what doctors can and cannot prescribe. These are clinical decisions reserved to individual practitioners. Licensing was introduced in the wake of the thalidomide disaster of the 1960s and is about the claims manufacturers are permitted to make in pursuit of sales. For pharmaceutical companies, licensing is a commercial imperative, the mechanism that allows them to, in effect, advertise their wares. Just because a product has achieved a licence for an indication does not mean it is the only compound endowed with the necessary benefits. It simply means that the manufacturer has invested in the studies to justify promoting that drug for that indication. It is inevitable, therefore, that doctors hear a lot about ‘new’ licensed indications, for telling us was the point of all that expenditure in creating the script. This clamour does not allow us to infer that the story is either specific to their product or previously untold.
The clinical diversity of antipsychotics was evident from the start, with a number being advocated for anxiety and depression rather than – or in addition to – schizophrenia, as well as non-psychiatric indications. Furthermore, by the 1980s it was difficult to undertake studies on bipolar disorder treated with lithium alone, for clinical practice relied heavily on combined regimes of lithium and antipsychotics.
Antipsychotics licensed for use in mood disorders may reflect new drug actions. More probably, this simply formalises evidentially what was clinical wisdom before – that (some) antipsychotics beneficially affect mood.
Principles
There are no specific ‘rules’ for prescribing older antipsychotics that should not apply to the newer drugs also, though the formulations and marketing materials for newer drugs make it easier to suspend the decision-making ‘algorithms’ that should be exercised in making conscious choices. Prescribing has become all too mechanical.
For exam purposes, trainees tend to organise antipsychotics in terms of chemical groupings, although this was not the traditional emphasis. Initially, clinical considerations predominated (Reference EllenbroekEllenbroek 1993). Europeans dissented from US psychiatrists in believing that antipsychotics were fundamentally different and could be classified along various efficacy/tolerability axes, though none of the often intricate systems proposed (e.g. the star scheme of Reference Bobon, Bobon and PinchardBobon et al (1972) and the multi-focal scheme of Reference Fischer-Cornelsson, Ferner and SteinerFisher-Cornelsson et al (1974)), achieved general acceptance. Most clinicians would now accept the American view that in terms of efficacy (and, post-CATIE, effectiveness too) all antipsychotics are comparable – though this is not to say that they achieve ‘end-point’ comparability by similar means. The Europeans were not necessarily wrong either. Chemical classifications can be helpful – but only in so far as they allow one to infer other, more clinically relevant characteristics about how a particular compound achieves its effects and at what cost. Two useful parameters are potency and breadth of receptor binding profile.
Potency, dosing and dose equivalence
Potency is another way of considering dose – higher-dosed compounds are in general of lower potency (see below for how pharmacokinetic variables can affect this principle). There are no clear-cut boundaries dividing compounds on the basis of potency, but a rough guide is that drugs with antipsychotic efficacy in units of milligram dosage per day are high potency; those with doses in tens of milligrams per day are intermediate; those where efficacy requires hundreds of milligrams per day are low potency.
Proper dose-finding studies were rarely done on early compounds and, with its consistently favourable therapeutic index, chlorpromazine became subject to relentless dose escalation soon after it arrived in North America (Reference HealyHealy 1996), a process repeated with all the older antipsychotics. One of the positive influences to flow from the development of newer antipsychotics has been the imprinting on prescribers of trial-based boundaries to dosing that hardly exist for older drugs. The importance of this difference between new and older antipsychotics to prescribing ‘consciousness’ cannot be overestimated. Trial-derived schedules endorsed by being licensed breed conservatism, a sense of moderation beneath a clearly defined dose ceiling. Perception could not be more different for older drugs, where dose recommendations arise for the most part from (bad) practice and repute, are seen to be infinitely flexible and beneath a ceiling that is barely evident.
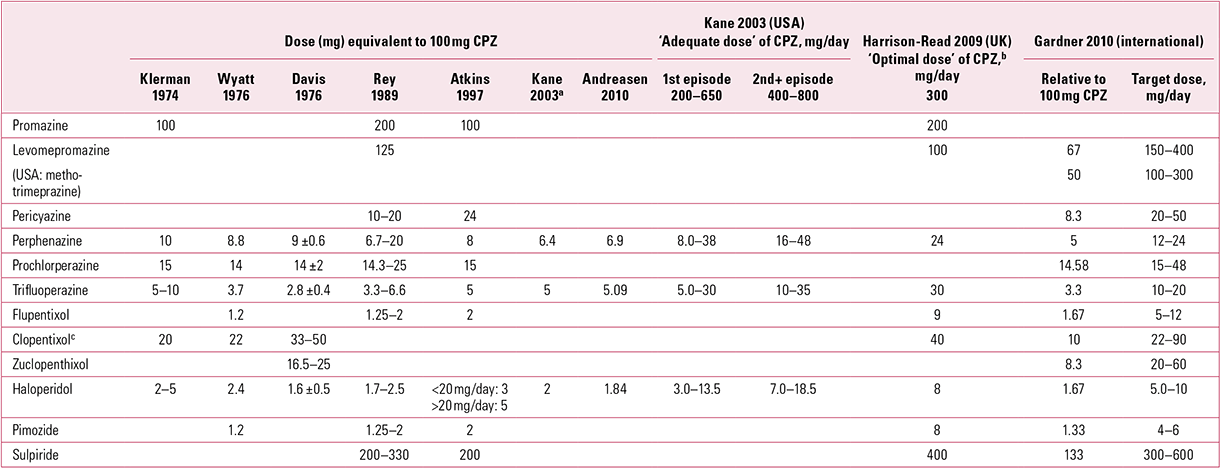
Even where dose-finding data are available, attempts to produce relative dose equivalences have come up with little more than a general consensus (e.g. Reference DavisDavis 1976; Reference WyattWyatt 1976; Reference Rey, Schulz and CostaRey 1989; Reference Atkins, Burgess and BottomleyAtkins 1997; Reference Kane, Leucht and CarpenterKane 2003; Reference WoodsWoods 2003; Reference Gardner, Murphy and O'DonnellGardner 2010). There are many reasons for this, including the inherent variability of patient responses, the prescribing culture (e.g. varying conceptions of what is therapeutic in different parts of the world, at different timesFootnote a) and the complex and varied pharmacokinetics these efforts cannot take into account. Furthermore, equivalences estimated by experts must have greater credibility if expertise has been gained in the clinic rather than (or in addition to) the library, as the latter type of expertise risks perpetuating past misconceptions. Even sophisticated approaches to equivalence involving calculation of dose–response curves from fixed-dose randomised controlled trials have to acknowledge the influence of an ‘uncertainty principle’ (Reference Davis and ChenDavis 2004).
Dose equivalence has repeatedly been shown to be least accurate with the high-potency older drugs, which are accordingly administered in substantially greater equivalent doses than low-potency compounds (Reference Baldessarini, Katz and CottonBaldessarini 1984; Reference Dewan and KossDewan 1995). An obvious reason for this is that low-potency drugs set their own dosage ceiling by virtue of anti-autonomic and other ‘non-target’ actions.
Concepts of dose equivalence should never be seen as absolute. Importantly, tables of comparison (e.g. Table 1) are best thought of as presenting the median points of ranges of dosing within which the therapeutic effect is most likely to be manifest, with the clinician’s aim always being to seek the minimum effective dose (see Reference OwensOwens 2012, this issue).
TABLE 1 Older (‘first-generation’) antipsychotics: some suggested dose equivalences to chlorpromazine (CPZ)
Breadth of receptor binding profile
Receptor binding profile allows inferences about, first, the extent to which a drug is likely to be endowed with non-extrapyramidal (general) adverse effects that bear on safety and tolerability, and second, what the likelihood might be of it possessing a built-in mechanism against the antidopaminergic actions it inevitably promotes (if one accepts the major inference of Laborit’s observations, noted above), the most important of which is Parkinsonism, including its subjective component. The default position for clinicians should be that all antipsychotics have the potential to produce Parkinsonian features in a dose-dependent fashion – a potential modifiable by individual susceptibility (i.e. dopamine ‘endowment’) and whatever anti-Parkinsonian actions the drug exerts.
Again, however, clinicians should not view receptor binding profiles as absolute, as data vary from study to study, depending on methodology, sample studied, species, and so on. A further point is that binding profiles derived in the laboratory can project poorly onto clinical scenarios – for example, they do not take account of the actions of metabolites, many of which are active and have their own kinetic and dynamic properties. Also, the binding profiles advocated so prominently concentrate on pathways long open to study – quite simply, those that can be readily investigated (relating predominantly to catecholamines, indoleamines, histamine and acetylcholine). The fact that these seem relevant to mode of action and tolerability is, one might say, a happy coincidence. As a diverse group of reactive compounds, however, antipsychotics interact with a wide range of proteins, including receptor types beyond the commonly studied (e.g. glutamatergic, sigmoid and ion channels), transporters (Reference Tatsumi, Jansen and BlakelyTatsumi 1999) and variants of known structures whose roles remain to be delineated (e.g. 5-HT6 and 5-HT7).
With these many cautions and caveats, pie charts (Fig. 1) can nonetheless provide at-a-glance impressions of relative profiles to guide, more than determine, treatment choices. Likewise, recommendations for each drug can be presented in outline (as in the Tables 2, 3 and 4 below), though they represent no more than proposals based on the overriding principle of minimum effective dose (‘start low, rise slow’). Confidence in choices as well as patterns of usage will only come with experience.

FIG 1 Receptor binding profiles of some of the phenothiazine antipsychotics, showing percentage of total binding contributed to by each transmitter type (after Reference Hyttel, Larsen, Christensen, Casey, Chase and ChristensenHyttel 1985).
TABLE 2 Older antipsychotics for oral use: aliphatic phenothiazines
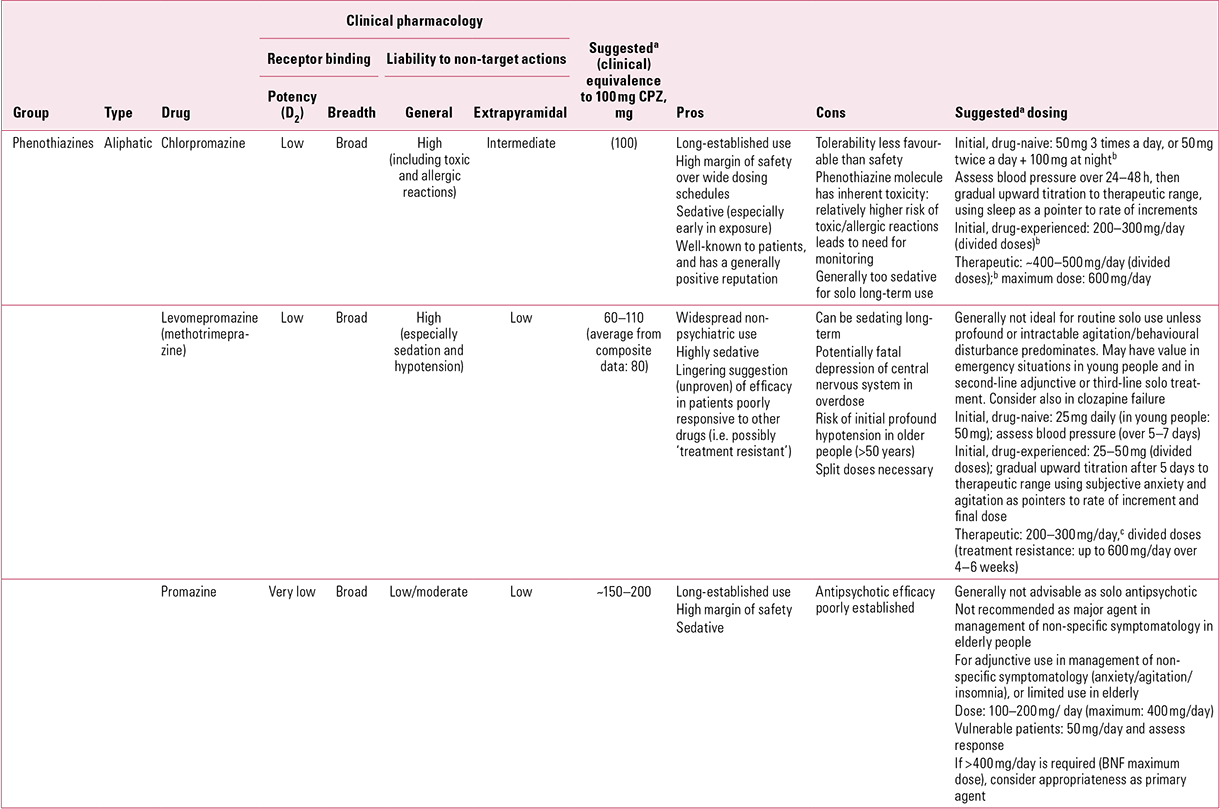
TABLE 3 Older antipsychotics for oral use: piperidine phenothiazines

TABLE 4 Older antipsychotics for oral use: piperazine phenothiazines

Phenothiazines
The substituted phenothiazines comprise a formidable group of over 40 compounds available around the world as anti-nausea/sickness treatments (especially vestibular-mediated forms associated with vertigo and with travel), antihistamines and drugs for post-operative and other forms of pain control, as well as antidepressants and anti-anxiety agents. Many more are in research use. Only a small proportion of the many synthesised survive as antipsychotics.
As psychiatry has become indifferent to the charms of phenothiazines, other disciplines have found excitement in their multiplicity of actions (Reference Motohashi, Kawase and SatohMotohashi 2006; Reference Sudeshna and ParimalSudeshna 2010). They have, for example, antimicrobial effects against bacteria (including mycobacterium tuberculosis), certain protozoa and fungi, and even prions, and they antagonise the calcium-controlling protein calmodulin, one of a number of possible ways in which they can facilitate cell death (Reference MotohashiMotohashi 1991). The potential of these multiple actions in anti-cancer therapy is under active investigation. This gives legs to a long-running debate – do patients with schizophrenia have lower rates of certain cancers? The question is difficult to address and results (over decades) have been contradictory. However, if general confusion does hide a specific negative association, the ‘hidden’ pharmacology of phenothiazines might be one explanatory contender.
Chlorpromazine (Table 2)
The original antipsychotic, chlorpromazine is a drug that trainees will still be familiar with, for its utility in clinical practice is such that even the onslaught of ‘atypicality’ has failed to extinguish its entrenched position, in Europe at least.
In vitro, chlorpromazine has moderate affinity for dopamine D2 receptors across the board (Fig. 1), though affinity is greatest for the specific D2 sub-type. Although this might suggest intermediate, not low, clinical potency, the paradox is probably explained by the drug’s low bioavailability (~30%), the result of extensive distribution (compare with sulpiride in Reference OwensOwens 2012, this issue). With other D2 family subtypes and with D1 receptors, chlorpromazine binds with comparable affinity, though this is less than it is for the specific D2 subtype (for representative values, see Reference Owens, Johnstone, Owens and LawrieOwens 2010).
With acute usage, the most striking effect relates to central H1 (antihistaminic) actions, as chlorpromazine is sedating on first exposure. This is a dose-related phenomenon that was not striking with the single low doses employed early in its development, as noted above, but is virtually universal with those now utilised in psychiatric practice (>100 mg). This action is unsurprising, for central antihistaminic activity was what was being sought when the compound was synthesised and was what selected it for development (Reference SwazeySwazey 1974). In addition to feeling sedated, chlorpromazine-naive patients may sleep for protracted periods, but these potentially useful early effects tend to habituate rapidly (over a 48–72 h period). Re-emergence of sedation in the post-acute phase of management suggests resolution of the underlying illness process and is an indication for cautious dose reductions. Persistence of low-grade sedation (as opposed to apathy) is the major reason why chlorpromazine on its own is nowadays of limited use as a long-term maintenance agent.
Chlorpromazine is also a powerful antagonist of α1 noradrenergic receptors (significantly more so than at D2 sites), which points to the major constraint on its unbridled use in acute situations – a potent hypotensive action. This is mainly postural but early in exposure can affect supine blood pressure also, necessitating supportive measures: bed-rest and/or raising the foot of the bed are usually sufficient. This also tends to fairly rapid habituation but can result in dramatic declines in blood pressure, especially following parenteral administration. For this reason, chlorpromazine should never be administered intravenously and even the intramuscular route, previously popular, has fallen from favour with the identification of safer alternative drugs.
Relatively weaker activity at muscarinic cholinergic receptors (Reference Owens, Johnstone, Owens and LawrieOwens 2010) predicts a modest anticholinergic adverse effect profile, in keeping with the greater tendency of chlorpromazine than some other low-potency phenothiazines to promote ‘acute-end’ extrapyramidal side-effects (EPS; i.e. acute dystonias, akathisia and Parkinsonism). It is also a potent antagonist at serotonergic 5-HT2A sites, as well as having lesser effects on 5-HT2C and especially 5-HT1A. In other words, this ‘typical’ antipsychotic is typically ‘atypical’!
Promazine (Table 2)
It was known early that addition of electronegative moities at position 2 (the R1 substitution) enhanced the antipsychotic potency of phenothiazines. Thus, promazine, the dechlorinated precursor of chlorpromazine, despite possessing the same general receptor binding profile, would be expected to be less potent. Early clinical comparisons confirmed this, with the same doses as with chlorpromazine providing no significant response (Reference Fleming, Spencer and WhitelawFleming 1959). However, some authors suggested that potency might be only slightly diminished, with Reference Azima and DurostAzima & Durost (1957) reporting that an average daily increase of just 25% resulted in improvements similar to those with chlorpromazine. Reference ReesRees (1960) estimated a chlorpromazine:promazine ratio of 1:3, while Reference Rey, Schulz and CostaRey et al (1989) calculated an overall potency of 1:2, which tends to reflect a longstanding consensus.
Early studies also tended to the conclusion that even in higher doses, promazine was better tolerated than chlorpromazine, with a low risk of jaundice (Reference Azima and DurostAzima 1957) and an especially low risk of EPS and postural hypotension (Reference Azima and DurostAzima 1957; Reference FrainFrain 1957; Reference Simpson and JessonSimpson 1958).
In most early studies, which assumed equipotency or only slightly diminished potency, promazine fared badly (Reference Mangun and WebbMangun 1956). It has therefore come to be seen as a ‘bland’ drug, best suited to those with particularly poor tolerability – especially the elderly – or, in low dose, as a general anti-anxiety agent for one-off or short-term use. Whatever its true comparative ‘anti-psychosis’ potency, this view is unlikely to shift. Any favourable appraisal of tolerability must, however, take account of the fact that promazine is a phenothiazine, so may, in the elderly, increase the liability to cerebrovascular events (see Reference OwensOwens 2012, this issue).
Levomepromazine (methotrimeprazine) (Table 2)
This is the only psychotropic with two official names, though outside the USA the former is now preferred. Developed by the same team behind chlorpromazine, it was introduced in 1956, but French physicians could not settle on a precise indication. It was advocated for depressive states and for ‘senile’ patients, as well as for schizophrenia, particularly for those who were ‘restless’, indications which reflect its sedative profile. It also caught the eye of anaesthetists as a useful pre-operative sedative, with efficacy in managing nausea and post-operative and other forms of pain. And it alleviated bronchoconstriction!
In the UK, non-psychiatric uses (e.g. surgery, palliative care) have predominated and levomepromazine has struggled to find a substantial psychiatric market. It is not, however, a drug without interest. Its sedative profile makes it potentially useful in psychiatric intensive care, and from the start it acquired a reputation in ‘treatment-resistant’Footnote b schizophrenia (Reference Payne and VerinderPayne 1960) that has never entirely faded (Reference Lal, Thavundayil and NairLal 2006). Whether this represents an effect independent of its sedative actions remains unclear.
Levomepromazine has a broad receptor binding profile (Fig. 1), covering antagonist actions at D1–D4, 5-HT1 and 5-HT2, noradrenergic (including, unlike chlorpromazine, α2 as well as α1), histamine H1, and muscarinic M1 and M2 sites (Reference Lal, Nair and CecyreLal 1993). Clinical impression is of a drug that is more sedative than chlorpromazine. Indeed, levomepromazine may be the most powerfully sedative of all antipsychotics, perhaps related in this case to the potency of its anti-noradrenergic as opposed to its antihistaminic actions. Noradrenergic antagonism is also likely to be responsible for the tendency to hypotension, which can be profound. Its EPS liability seems somewhat, though not strikingly, less than chlorpromazine (Reference Lal, Thavundayil and NairLal 2006).
Although generally considered a low-potency drug, the ideal levomepromazine dose regime for treating psychotic illness was never well established and experience remains scant. While 300 mg/day was not found to have efficacy (Reference Milne and FowlerMilne 1960), 600 mg/day or more was (Reference Payne and VerinderPayne 1960; Reference Lal and NairLal 1992, Reference Lal, Thavundayil and Nair2006). This may suggest equipotency with chlorpromazine, but most of these studies were in institutionalised patients with severe or treatment-resistant schizophrenia. In general, greater potency is usually accepted (Reference BanBan 1971), with figures varying between twice that of chlorpromazine and equipotency (Reference ReesRees 1960; Reference Rey, Schulz and CostaRey 1989; Reference Gardner, Murphy and O'DonnellGardner 2010). Composite estimates (e.g. from Reference Rey, Schulz and CostaRey 1989 and Reference Gardner, Murphy and O'DonnellGardner 2010) do, however, suggest that the increase may only be slight, with a chlorpromazine:levomepromazine ratio of around 100:80. This is in line with the lower optimal maintenance doses (50–100 mg/day) suggested earlier (Reference Payne and VerinderPayne 1960). The British National Formulary (BNF) upper recommended dose of 1 g/day remains more in line with the perception of equipotency and is undoubtedly on the high side (British Medical Association 2012).
Levomepromazine must be used with care in the elderly and others in whom hypotension could have serious consequences and, in line with its sedative effects, in those at risk of suicide, as it has been associated with higher toxicity and death rates following overdose than most antipsychotics (Reference Schreinzer, Frey and StimpflSchreinzer 2001). Although a powerful inhibitor of the cytochrome P450 2D6 (CYP2D6) enzyme system, this does not appear to cause significant problems in the antipsychotic combinations studied (such as risperidone; Reference Yoshimura, Shinkai and KakiharaYoshimura 2005), but it may produce a significant impairment of the O-demethylation of codeine to morphine, rendering codeine-based analgesics less effective (Reference Vevelstad, Pettersen and TallaksenVevelstad 2009). It is also possible that induction of CYP1A2 or CYP3A3/4 systems underlie a lowering of clozapine levels (Reference Bugamelli, Mandrioli and KenndlerBugamelli 2007).
Psychiatry in the UK has probably unduly neglected levomepromazine as a tool in the management of marked and persistent behavioural disturbance or in adjunctive treatment of non-specific symptomatology (such as anxiety and insomnia). Its potential in treatment resistance, operationally defined or otherwise, requires further study, but this may be a drug worth considering in those for whom, by virtue of clozapine failure, few other options remain.
Pericyazine (periciazine) (Table 3)
Pericyazine also followed chlorpromazine from Rhône-Poulenc’s laboratories, a decade later but accrued, largely post hoc, the sort of ‘panacea’ indications that foster scepticism – ‘character disorders’, ‘psychopathy’, anxiety states and organic disorders, as well as schizophrenia (Anonymous 1967). The likely common factor related to its benefits in situations, as opposed to disorders, characterised by overactive, confrontational or aggressive behaviour. With such an ill-sketched profile and life in the shadow of its elder sibling, it is hardly surprising that pericyazine has held on to a tenuous place in clinical practice and is little known, far less used, in the UK, where awareness stems largely from proceedings of a single sponsored symposium (Reference Jenner, Hays and FlynnJenner 1965). However, pericyazine did enjoy popularity in Scandinavia, Russia and Japan.
No detailed in vitro receptor binding profile is available for pericyazine (Reference Fleming, Yao and RavikumarFleming 2010), though it is known to be more potent at α-adrenergic than dopamine sites and can therefore be associated with hypotension, especially on first exposure. It also has powerful antiserotonergic actions, the clinical significance of which is unclear. Its tolerability profile indicates anticholinergic-type actions, and with EPS liability probably comparable to chlorpromazine, this suggests a more potent action at muscarinic sites also. Reports of cardiac dysrhythmias, though rare, might suggest an ion channel action.
Such has been pericyazine’s disregard that it is not possible to be confident about the optimal antipsychotic dosage, far less its relative potency. It is certainly several times (4–20) more potent as an anti-emetic than chlorpromazine (Anonymous 1967). Other data, not published in peer-reviewed literature, suggest comparable antipsychotic efficacy from 15 mg/day pericyazine as from 600 mg/day thioridazine (Reference Heller, Mather, Jenner and HaysHeller 1965). However, Reference Barker and MillerBarker & Miller (1969), studying chronic schizophrenia, got comparable efficacy with a 1:10 ratio against thioridazine (approximately 1:9 against chlorpromazine). Taking these data together, Reference Rey, Schulz and CostaRey et al (1989) calculated a five- to tenfold greater potency. The BNF, on the other hand, seems to regard it as a sort of chlorpromazine-Mk 2, suggesting rough dose equivalence or only slightly enhanced potency (British Medical Association 2012).
Equivalence is not the clinical experience of most, which suggests that pericyazine is indeed in the order of four to ten times more potent as an antipsychotic than chlorpromazine (Atkins 1987; Reference Rey, Schulz and CostaRey 1989; Reference Gardner, Murphy and O'DonnellGardner 2010). Accounting for EPS liability, it would be prudent to go with the higher potency assumption to begin with (chlorpromazine:pericyazine ratio of 10:1) in order to minimise exposure.
Its most striking – and useful – characteristic is its powerful anti-autonomic and sedative effects, no doubt arising from central antihistaminic actions. The impression is that pericyazine, at ‘anti-psychosis’ equivalence, is another drug disproportionately more sedative than chlorpromazine. Again, this can be a disadvantage for solo, long-term use but for short-term, acute and, especially, adjunctive use (in the management of non-specific symptomatology) pericyazine has, in my view, practical value beyond what its current utilisation would suggest. This enhanced sedative role is likely to be why it has been found to be more effective in treating aggression and hostility than chlorpromazine (Reference BeckerBecker 1981) and why original indications became so blurred. It may also be why pericyazine is referred to surprisingly positively in patient-targeted online resources (Reference Matar, Almerie and MakhoulMatar 2008).
Since the withdrawal from the European market in 2002 of thioridazine, a drug well regarded by both patients and doctors, pericyazine is the sole remaining oral piperidine phenothiazine in the UK. It is, however, debatable whether the QTc prolongation issue would have sunk thioridazine quite so conclusively had the safety profiles of the second-generation antipsychotics with which it was in competition been as comprehensively understood then as they are now (see Reference OwensOwens 2012, this issue). Thioridazine and its metabolites do continue to stimulate interest as potentially cheap and safe antituberculous and antimalarial agents for poorer countries (Reference Amaral, Viveiros and KristiansenAmaral 2001; Reference ThanacoodyThanacoody 2007).
Perphenazine (Table 4)
Perphenazine was introduced in 1957 and, in addition to an international market for the oral formulation, is also available as depots (decanoate and enanthate) in several European countries. It has recently grown to prominence as the ‘standard’ comparator in CATIE (Reference Lieberman, Stroup and McEvoyLieberman 2005).
Perphenazine is active at 5-HT2 sites (Fig. 1) and has significant anti-α-adrenergic actions, promoting hypotension on first exposure, especially if administered parenterally, though this is delayed compared with most other antipsychotics (Reference Bhargava and ChandraBhargava 1964), suggesting the action of a metabolite. It is also sufficiently antihistaminic to have sedative properties – low doses (e.g. 2 mg) can produce drowsiness without hypotension, even when administered intramuscularly, giving it a safety margin over chlorpromazine (Reference Musey, Preedy and MuseyMusey 1986). It has no significant anticholinergic action (Reference Sweet, Pollock and MulsantSweet 2000; Reference Chew, Mulsant and PollockChew 2008) and although well known to promote EPS in higher doses, its relatively good EPS tolerability at low to medium doses probably reflects the fact that its major metabolite, N-dealkyl perphenazine, which with chronic use attains blood levels 1.5–2 times those of the parent drug, shows modest antimuscarinic (M1) binding (Reference Sweet, Pollock and MulsantSweet 2000). This might therefore be a drug to start and hold low in order to allow build-up of the metabolite in the expectation of an anti-EPS effect as doses rise.
Although often stated as being of comparable potency to haloperidol (Reference Hartung, Wada and LauxHartung 2005), perphenazine is best considered as of intermediate potency. Based on animal data, the originally suggested chlorpromazine:perphenazine potency of 5:1 or 6:1 (Reference ReesRees 1960) would now seem an underestimate, with clinical potency best considered at about 10–15 times that of chlorpromazine (chlorpromazine:perphenazine = 100:8 mg), as some estimated from the start (Reference DiMascio, Havens and KlermanDiMascio 1963a). The BNF reflects this more generous view, recommending an upper daily dose of 24 mg (British Medical Association 2012), though this may be overcautious. In the USA, traditional practice tended to a relative potency of between 8:1 and 9:1 (Reference WyattWyatt 1976), in line with which CATIE allowed up to 32 mg/day (Reference Lieberman, Stroup and McEvoyLieberman 2005). However, modern specialist opinion also seems to be veering towards the lower range (24 mg) (Reference Gardner, Murphy and O'DonnellGardner 2010).
Perphenazine has rather poor kinetic properties, in particular low bioavailability (Reference Hansen, Christensen and ElleyHansen 1976), probably reflecting first-pass effects. This, combined with an average half-life of 8–12 h, means that the drug is best suited to administration three times a day, though for practical reasons twice daily is usual. However, in cases of poor response, increasing the frequency should be considered before a change of drug. In a small percentage of ‘poor metabolisers’, half-life is extended to up to 20 h, and in those who experience undue side-effects, especially sedation, reducing the frequency may be more appropriate than reducing the dose.
Like many psychotropics, perphenazine is metabolised predominantly by the CYP2D6 system. Although resistant to induction, this system is prone to inhibition. Paroxetine is a potent inhibitor of CYP2D6 isoenzymes and dramatic increases in perphenazine blood levels (2–13 fold) have been reported with the addition of the antipsychotic to stable paroxetine treatment, resulting in clinically relevant deteriorations in tolerability (sedation, EPS, impaired psychomotor performance and memory) (Reference Ozdemir, Naranjo and HerrmannOzdemir 1997). This type of kinetic interaction is likely to be relevant to all antipsychotics with significant metabolism via CYP2D6 when combined with a potent inhibitor. On the other hand, perphenazine is itself a weak inhibitor of this system, on which tricyclic antidepressants also depend for metabolism. Combinations of perphenazine and tricyclics are associated with up to 70% increases in tricyclic levels (Reference Linnoila, George and GuthrieLinnoila 1982). This fact is made use of in combination preparations (e.g. with amitriptyline in Triptafen®), though concern about whether, even then, adequate antidepressant levels can be achieved has resulted in a decline in their use.
Trifluoperazine (Table 4)
Trifluoperazine was previously widely used in the UK, not least in low dose as an ‘anti-anxiety’ agent and in combinations (e.g with the monoamine oxidase inhibitor tranylcypromine, comprising Parstelin®) as an ‘antidepressant’.Footnote c It still enjoys international usage and is also a major research tool, as its inhibitory actions on calmodulin have long been known and the conformational changes it produces in the protein are to some extent characterised (Reference Feldkamp, O'Donnell and YuFeldkamp 2010).
Trifluoperazine possesses high affinity for D2 receptors but has low affinities at D1 and serotonergic sites and relatively weak anti-α1 (but no α2) adrenergic actions (Reference Huerta-Bahena, Villalobos-Molina and Garcia-SainzHuerta-Bahena 1983). It has little other receptor activity (Fig 1). Its predicted lack of effect on blood pressure and excellent general tolerability were confirmed early in healthy volunteers (Reference Di Mascio, Havens and KlermanDiMascio 1963b). Its potency, however, combined with little inherent antimuscarinic activity, means that EPS liability is high. Despite little anticholinergic action in vitro, its use may still be associated with dry mouth, blurred vision and so on (Reference Marques, Soares and Silva de LimaMarques 2004), symptoms usually attributed to muscarinic blockade. This may, however, reflect an unduly narrow interpretation of how such symptomatology arises or the activity of metabolites rather than the parent drug.
Although trifluoperazine can be sedative (Reference Marques, Soares and Silva de LimaMarques 2004), especially at high dose, clinical experience suggests that, compatible with its binding profile, it is one of the least sedative antipsychotics. Originally, much was made of its ‘alerting’ properties, particularly in long-term institutionalised patients (Reference Gwynne, Hundziak and KavtschitschGwynne 1962), something also inferred from improvements in psychomotor abilities and speed of performance (to some extent dose-related) reported in healthy volunteers (Reference DiMascio, Havens and KlermanDiMascio 1963a). Apart from possible presynaptic dopaminergic (autoreceptor) effects, its known pharmacology does not offer a ready basis for such actions, which tend not to be commented on nowadays. Psychomotor test findings may have reflected mild Parkinsonism as, paradoxically, people with Parkinsonism may show enhanced performance in forced-choice paradigms, but as the first non-sedating antipsychotic, it was probably this characteristic that, in contrast with other phenothiazines, stood out to be interpreted as ‘alerting’.
Early clinical research suggested trifluoperazine to be a moderate- to high-potency compound with treatment efficacy most frequently found in the 5–30 mg/day range (Reference Madgwick, McNeill and DriverMadgwick 1958; Reference Macdonald and WattsMacdonald 1959; Reference PaynePayne 1959; Reference Sarwer-Foner, Koranyi and MackaySarwer-Foner 1959), though maintenance regimes of less than 10 mg/day have also been found to be effective (Reference PaynePayne 1959). In the USA, it tended to be considered in the intermediate potency range (Reference WyattWyatt 1976) but in the UK, relative chlorpromazine:trifluoperazine potency is usually taken to be around 20:1 (Reference Owens, Johnstone, Owens and LawrieOwens 2010). However, as a drug relatively free from anti-autonomic side-effects and hence prone to early and potentially needless dose escalations, its true relative potency may be as high as 30:1 (Reference Rey, Schulz and CostaRey 1989; Reference Gardner, Murphy and O'DonnellGardner 2010), which is more in line with a calculated effective dose in the range 10–15 mg/day (Reference Davis and ChenDavis 2004). Unusually, the BNF offers no guidance on upper dose limits for trifluoperazine in schizophrenia (British Medical Association 2012), but the evidence suggests that, despite excellent tolerability, doses above 15–20 mg/day should be considered ‘high’.
For a drug so widely used for over half a century, it is truly astonishing how little is known about trifluoperazine’s pharmacokinetics. The only sure fact is that they are highly variable across individuals on both single and repeat administration (Reference Midha, Korchinski and VerbeeckMidha 1983, Reference Midha, Hawes and Hubbard1988). Its terminal phase half-life, at around 12 h with single dosing (Reference Midha, Korchinski and VerbeeckMidha 1983), suggests that it is barely suitable for once-daily dosing, though this may extend (to up to 30 h) with regular use. Perhaps because of this, the previously popular modified-release (or ‘spansule’) formulation has unfortunately now been withdrawn in the UK. With the standard formulation, cautious practice might still dictate twice-daily administration.
Prochlorperazine
Prochlorperazine has never found favour as an antipsychotic in the UK, despite being structurally very similar to trifluoperazine (chlorine substitution at the R1 position instead of trifluoromethyl). Even the team behind its development was more interested in its much greater potency as an anti-emetic compared with chlorpromazine and its effects on migraine and vertigo (Reference ReesRees 1960), actions that have sustained its market position since.
Prochlorperazine was, however, initially greeted with enthusiasm by some psychiatrists. A recurrent theme in the early literature was that chlorpromazine offered mainly non-specific behavioural advantages, whereas prochlorperazine had a more favourable impact on fundamental domains of illness such as thought disturbances (Reference DenhamDenham 1958; Reference Wilson, McKay and SandiferWilson 1961) and affect (Reference Milne and BerlinerMilne 1958). Such observations probably reflected simply the conservative chlorpromazine doses at the time, especially in the UK, rather than any differential efficacy. It was also noted early that prochlorperazine had a greater liability to promote EPS than chlorpromazine (Reference DransfieldDransfield 1958) – which actually fitted the prevailing view that EPS were an inherent part of ‘anti-psychosis’ and hence a sign of enhanced efficacy!
Prochlorperazine has actions at dopaminergic, muscarinic and α-adrenergic sites, evident on single dosing so probably reflecting the profile of the parent drug. However, antihistaminic effects are mainly evident with repeat dosing, suggesting the actions of a metabolite (Reference Isah, Rawlins and BatemanIsah 1991). The resultant lack of sedative action with single dosing no doubt facilitated its approval in over-the-counter preparations for nausea/vomiting. In vitro, prochlorperazine is also active across a range of 5-HT subtypes, including 5-HT2 (Reference Roth, Craigo and ChoudharyRoth 1994), though strangely this is one antipsychotic that never seems to have garnered an ‘atypical’ plaudit.
Prochlorperazine’s R1 substitution would indicate lower potency than its close piperazine relative, trifluoperazine, and this is clinical experience, but a second factor (in addition to limited usage) that makes equivalence difficult to determine is the drug’s unfavourable kinetics. Prochlorperazine is subject to substantial first-pass effects and is very extensively cleared, resulting in bioavailability of only some 12.5% (Reference Isah, Rawlins and BatemanIsah 1991), low even by the standards of the highly lipophilic phenothiazines. This may be why, conventionally, it has been treated as of intermediate, veering to low, potency (unusual for a piperazine compound).
The early literature suggested chlorpromazine: prochlorperazine ratios between 2:1 (Reference DransfieldDransfield 1958) and 5:1 (Reference Wilson, McKay and SandiferWilson 1961), whereas Reference WyattWyatt (1976) calculated a relative potency of around 7:1, at the upper range of the 4:1 to 7:1 suggested by others (Reference Rey, Schulz and CostaRey 1989). On the other hand, Reference Wilson, McKay and SandiferWilson et al (1961) found that patients on 30 mg/day did better than those on 90 mg/day, which is in line with the higher potency (~14:1) assessed more recently, albeit with the low confidence of a drug seldom used by the experts (Reference Gardner, Murphy and O'DonnellGardner 2010).
Thus, while there is a suggestion that efficacy may be achieved with doses lower than the 75–100 mg/day recommended in the BNF (British Medical Association 2012), such unclear dosage parameters and half a century of neglect make it unlikely that prochlorperazine will now find the favour psychiatry has hitherto denied it. It would, however, be interesting to know how this generally well-tolerated drug performed as a low-dose antipsychotic in preparations formulated for buccal absorption (e.g. Buccastem®), which avoid first-pass effects and achieve blood levels twice those of oral formulations (Reference Finn, Collins and VoyksnerFinn 2005).
The rest of the family
My second article examines non-phenothiazines and discusses issues in the prescribing of ‘first-generation’ antipsychotics (Reference OwensOwens 2012, this issue).
MCQs
Select the single best option for each question stem
-
1 In the context of drug regulation, licensing implies:
-
a that a drug is safer than other members of its class which are unlicensed
-
b that a drug is more efficacious than other members of its class which are unlicensed
-
c that a manufacturer has provided sufficient data to allow for a favourable risk:benefit appraisal
-
d that a manufacturer has been granted copyright over the branded name of the product
-
e that only the licensed product can be prescribed for a particular indication.
-
-
2 Antihistaminic properties associated with the use of which one of the following are likely to reflect the actions of a metabolite?
-
a chlorpromazine
-
b pericyazine
-
c perphenazine
-
d prochlorperazine
-
e trifluoperazine.
-
-
3 Overdose of which one of the following is most likely to be associated with fatal respiratory depression?
-
a levomepromazine
-
b pericyazine
-
c promazine
-
d prochlorperazine
-
e thioridazine.
-
-
4 Of the following, the greatest first-pass effects are associated with:
-
a chlorpromazine
-
b levomepromazine
-
c pericyazine
-
d perphenazine
-
e trifluoperazine.
-
-
5 Dose equivalence estimates for antipsychotics are not contaminated by:
-
a actions of metabolites
-
b drug potency
-
c industry sponsorship
-
d when efficacy studies were undertaken
-
e where efficacy studies were undertaken.
-
MCQ answers

| 1 | c | 2 | d | 3 | a | 4 | d | 5 | c |






eLetters
No eLetters have been published for this article.