



Introduction
The application of biomaterials based on natural or synthetic polymers in implants accompanied the tremendous development in medicine within the last few decades. Modern clinical applications like regenerative therapies or minimally invasive surgical procedures require medical devices which are highly multifunctional.[Reference Liang, Li, Scott and Kiick1] Implants should modulate the immune response and degrade in a controlled manner or execute movements on demand or release a drug with defined kinetics.[Reference Taraballi, Sushnitha, Tsao, Bauza, Liverani, Shi and Tasciotti2–Reference Radke, Jia, Sharma, Fena, Wang, Goldman and Zhao4] For example, polyesters were designed for adjusting the degradation rate whereas polyamides were considered with regard to their high toughness and tensile strength, electrical insulation, heat- or abrasion resistance.[Reference Winnacker and Rieger5, Reference Brannigan and Dove6] Poly(ester amide)s contain ester and amide bonds and serve as platform for the development of highly sophisticated multifunctional biomaterials.[Reference Fonseca, Gil and Simões7] Poly- or even oligodepsipeptides (ODPs), which are prominent examples for poly(esteramide)s, are gaining an increasing impact as controlled drug delivery systems, as gene carriers and as scaffolds for tissue regeneration.[Reference Ghosal, Latha and Thomas8–Reference Feng and Guo10] From natural sources like marine invertebrates and cyanobacteria mainly cyclic ODPs are isolated which exhibit antimicrobial or protease inhibition activities, or cytotoxicity against cancer cells.[Reference Batiste and Johnston11–Reference Liang, Zhao, He, Mu, Deng and Han14] While initially, poly(depsipeptide)s were synthesized via polycondensation of activated depsipeptides,[Reference Nissen, Gilon and Goodman15] the state of the art synthetic route is ring opening polymerization of 2,5-morpholine derivatives.[Reference Feng, Lu, Behl and Lendlein16] By variation of the side-chains, different functionalities are introduced, enabling to tailor the properties of the macromolecules, which can be used as telechelics in building blocks for biomedical applications.[Reference Feng and Guo10, Reference Naolou, Lendlein and Neffe17, Reference Peng, Behl, Zhang, Mazurek-Budzyńska, Feng and Lendlein18]
The ODP chains are able to form inter- and intramolecular H-bonds between the ester and amide linkages. Due to these strong interactions, the ODPs exhibit relatively high thermal transition temperatures. By introduction of a hexyl side-chain in the α-hydroxy acid part of the molecules, the thermomechanical properties of the ODPs can be influenced. The atactic bulky side group impedes the crystallization, resulting in amorphous polymers. H-bonding between chain segments in the amorphous polymer is also hindered by the hexyl side-chains, resulting in a glass transition close to human body temperature.[Reference Peng, Behl, Zhang, Mazurek-Budzyńska, Razzaq and Lendlein19] Telechelic ODPs are used for the preparation of (multi)block copolymers with biodegradability,[Reference Elomaa, Kang, Seppälä and Yang20–Reference Peng, Behl, Zhang, Mazurek-Budzyńska, Feng and Lendlein23] shape-memory properties,[Reference Feng, Behl, Kelch and Lendlein24] or particle forming capabilities required for building drug carrier systems.[Reference Zhang, Feng, Tian, Zhao, Khan and Guo25–Reference Brunacci, Wischke, Naolou, Neffe and Lendlein27] Nanoparticles have been prepared from di- and triblock copolymers using hydroxyl mono- and difunctional oligo(3-(S)-sec-butylmorpholine-2,5-dione) (OSBMD) and linear polyethylenimine. The large hydrophobic side-chains were chosen to stabilize the secondary structures, which were hypothesized to contribute to particle stability.[Reference Wang, Naolou, Ma, Deng, Xu, Mansfeld, Wischke, Gossen, Neffe and Lendlein28] Thermoplastic multiblock copolymers prepared from oligo(3-(S)-iso-butylmorpholine-2,5-dione)-diol (OIBMD-diol) and oligo(ε-caprolactone)-diol (OCL) segments exhibit reversible actuation.[Reference Yan, Rudolph, Noechel, Gould, Behl, Kratz and Lendlein29] Moreover, materials based on multiblock copolymers of OCL and OIBMD (OCL-OIBMD) are consulted for the enhanced proliferation of endothelial cells.[Reference Bai, Li, Duo, Hao, Zhang, Shi, Guo, Ren and Feng30] Electrospun scaffolds are prepared using alternatively layer-by-layer electro-spinning and electrospraying techniques of OCL-OIBMD and silk fibroin, which encapsulate plasmid complexes.[Reference Feng, Liu, Ren, Lu, Guo, Behl, Lendlein and Zhang31]
For many poly(depsipeptide)s mentioned here, the direct interaction of the material with a biological environment, e.g., cells,[Reference Knight, Stutchbury, Imruck, Halfpap, Lin, Langbein, Gillies, Mittler and Mequanint32] is crucial for success in application. Nevertheless, it is still challenging to control the complex interplay of the bulk properties (stability; mechanical properties) and the variety of interactions of the material surface with the biological environment, especially with proteins from the extracellular matrix or with cell membranes. Therefore, a detailed knowledge about the interfacial behavior of ODPs under different environmental conditions is highly demanded. In our approach to elucidate the interfacial behavior of ODPs, we use Langmuir monolayers at the air–water interface as suitable models for evaluating the material surface behavior in a physiological environment.[Reference Schone, Roch, Schulz and Lendlein33] Here, we investigate the interfacial behavior of three telechelic ODPs, namely: OMMD: oligo(3-methyl-morpholine-2,5-dione)diol, OIBMD: oligo[3-(S)-iso-butylmorpholine-2,5-dione]diol, and OSBMD: oligo[3-(S)-sec-butylmorpholine-2,5-dione]diol as well as a multiblock copolymer containing nearly equal amounts of OCL and OIBMD (OCL-OIBMD) (Scheme 1). These depsipeptides are chosen because their complete hydrolysis produces glycolic acid and amino acids. The methyl side-chain amino acid (alanine) as well as glycolic acid are normal body metabolites[Reference Ashammakhi and Rokkanen34, Reference Bier, Arnold, Sherman, Holland, Holmes and Kipnis35] whereas the butyl side-chained amino acids (leucine and iso-leucine) are essential. We therefore expect excellent metabolization of these depsipeptides when used for drug delivery or as implant material. The side-chain length is varied to study the influence on secondary structure, crystallization, and thermal transitions. The behavior of OCL-OIBMD multiblock copolymers is studied because of their prospective application as shape-memory material with switching temperature around 37 °C. In a first step, the interfacial activities and stabilities of the different oligomers and the multiblock copolymer were compared. The interfacial activity is related to the amphilicity of the molecules, which in turn determines their ability to interact with cell membranes. Since the ODPs are highly regular molecules capable of hydrogen bonding, it seems probable that these molecules can self-assemble into supramolecular structures similar to polypeptides like poly(γ-benzyl-glutamate).[Reference Niehoff, Mantion, McAloney, Huber, Falkenhagen, Goh, Thünemann, Winnik and Menzel36] Compared to bulk systems, self-assembled structures can be much more easily formed and observed in monolayers. Therefore, we applied Brewster angle microscopy (BAM) and polarization modulation infrared reflection absorption spectroscopy (PM-IRRAS) to monitor the arrangement of the polymer chains at the air–water interface. PM-IRRAS provided information about the orientation and the conformation at the molecular level allowing a statement about the secondary structure, which is especially interesting for depsipeptides that are expected to assume secondary structures differing from peptides. To further investigate the organization of ODPs, monolayers were transferred to solid substrates and studied via atomic force microscopy (AFM). Since OCL-OIBMD is a prospective resorbable biomaterial, the effect of multiblock copolymer formation on the degradation behavior of OCL and OIBMD was also evaluated.
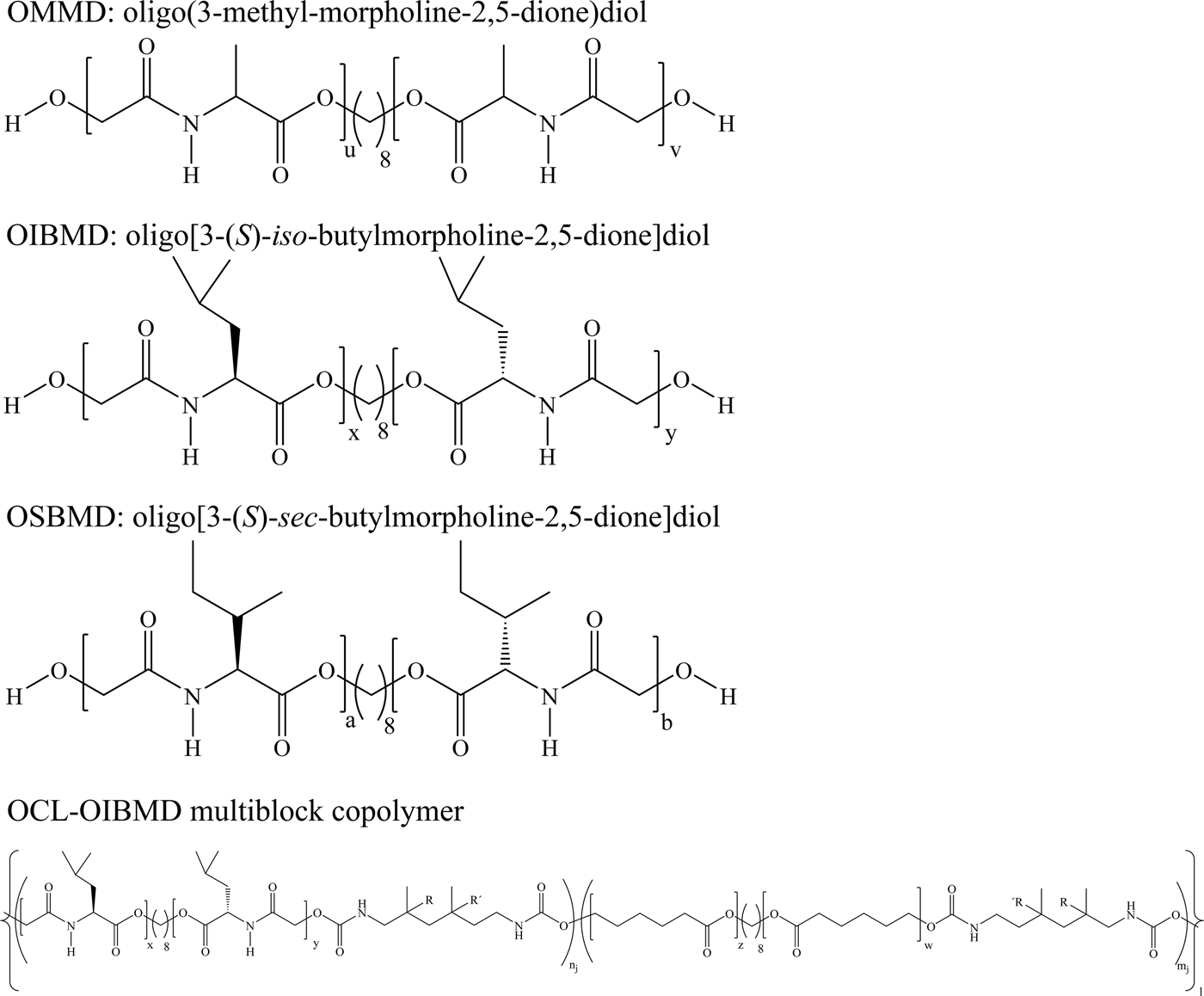
Scheme 1. Chemical structures of the investigated ODPs and the investigated multiblock copolymer. Here, (u, v) and (x, y) and (a, b) are the numbers of depsipeptide repeat units in the three different oligomers. The sums (x + y) and (a + b) and (u + v) are determined by the number average molecular weights of the corresponding ODPs via x + y = (M n−moctanediol)/mIBMD. Similarly, z and w are the numbers of ε-caprolactone repeat units where the sum of z and w is determined by the number average molecular weight of the oligo(ε-caprolactone)diols. R can be a proton or methyl group. If R is a proton, R′ is a methyl group and vice versa. The numbers n j and m j define the numbers of consecutive OIBMD or OCL blocks in each of the l OIBMD—OCL sequences. The roughly equal numbers of ε-caprolactone and depsipeptide units demand that 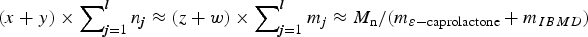 $(x + y) \times \sum\nolimits_{{\bi j} = 1}^{\bi l} {n_{\bi j}\approx (z + w)} \times \sum\nolimits_{{\bi j} = 1}^{\bi l} {m_j\approx M_{\rm n}/ \lpar m_{{\rm \varepsilon} -{\rm caprolactone}} + m_{IBMD} \rpar}$.
$(x + y) \times \sum\nolimits_{{\bi j} = 1}^{\bi l} {n_{\bi j}\approx (z + w)} \times \sum\nolimits_{{\bi j} = 1}^{\bi l} {m_j\approx M_{\rm n}/ \lpar m_{{\rm \varepsilon} -{\rm caprolactone}} + m_{IBMD} \rpar}$.
Materials and methods
Oligo[3-(S)-sec-butylmorpholine-2,5-dione]diol (OSBMD), oligo[3-(S)-iso-butylmorpholine-2,5-dione]diol (OIBMD), and oligo(3-methyl-morpholine-2,5-dione)diol (OMMD) were synthesized via ring opening polymerization according to Refs. Reference Feng and Guo10 and Reference Feng, Behl, Kelch and Lendlein24. The block copolymers with a molecular weight of M n = 43,000 g/mol were synthesized by joining equimolar amounts of oligo(ε-caprolactone)diols and OIBMD blocks with trimethyl-1,6-diisocyanatohexane according the procedure in Ref. Reference Feng, Behl, Kelch and Lendlein24. Structural characterization by NMR (see Fig. S1 in Supplementary Material) confirmed that the block copolymer contained nearly equimolar amounts of OIBMD and oligo(ε-caprolactone) monomers.
Number-average molecular weights (M n) and weight-average molecular weights (M w) were determined using three different methods: 1H-NMR, Gel Permeation Chromatography (GPC), and OH-group titration. 1H-NMR measurements were performed at 25 °C in DMSO-d6 using a Bruker Avance II 500 spectrometer (500 MHz). The procedure and formulas used to calculate the molecular weight of the ODPs are given in Ref. Reference Feng, Behl, Kelch and Lendlein24.
GPC was performed using 50 mmol ammonium acetate in N,N-dimethylformamide at 35 °C as an eluent with a flow rate of 0.25 mL/min. As internal standard 0.005 wt% 2,6-di-tert-butyl-4-methylphenol was used for all samples. The system was equipped with 250 mm × 4.6 mm GRAM gel columns and 3 × 102 nm porosity, 10-μm particle size (Polymer Standard-Service GmbH, Mainz, Germany, polystyrene standards (PSS)), a degasser (ERC-3315, Riemerling, Germany), a gradient pump PU 980, and an automatic injector AS-851 (both Jasco, Tokyo, Japan). Three detectors were used: a multi wavelength detector MD-910 (270 nm), a refractive index detector RI-930 (both Jasco), and the differential viscometer n-1001 (WGE, Dr. Bures, Dallgow, Germany), which were combined by a split. All molecular weights were determined using a universal calibration with PSS.
The molecular weight of the diols was also determined via OH-titration of the macromolecules dissolved in N,N-dimethylformamide (DMF) (Merck, Darmstadt, Germany) with 0.1 N-tetrabutylammonium hydroxide (Merck, Darmstadt, Germany). The titrations were performed with the titrator DMS Titrino 716 (Metrohm, Zofingen, Switzerland) and a solvotrode with LiCl (sat.) in ethanol. The determination of the hydroxyl groups is based on the esterification of the OH-groups with acetic anhydride (abundantly, Sigma-Aldrich, St. Louis, MO, USA) in the presence of N-methylimidazole (Sigma-Aldrich, St. Louis, MO, USA) as a catalyst at ambient temperature and by back titration.
Langmuir film balance
The surface pressure–area (π–A) isotherms were recorded at 22.0 ± 1.0 °C with a Langmuir high-compression trough (KSV NIMA, Finland) placed on an active vibration isolation system (halcyonics variobasic 40, Accurion, Göttingen, Germany) within a laser safety cabinet or a Langmuir–Blodgett trough type NIMA-622, respectively. The surface pressure (π) was measured with a calibrated sensor located equidistant from each barrier via the Wilhelmy technique. For Langmuir monolayer experiments the used water was purified by a Milli-Q Gradient A-10 water purification system (18.2 MΩ/cm, toc < 4 ppb, Millipore, Merck, Darmstadt, Germany). For the formation of monolayers, the sample solution was spread drop-wise onto the clean water surface using a 250 µL microsyringe (825 RN SYR, Hamilton Co., Reno, NV, USA). Chloroform was used as spreading solvent (HPLC grade, Carl Roth, Karlsruhe, Germany) for OIBMD and OSBMD. For OMMD monolayers, a 98:2 chloroform dimethylformamide mixture was used for spreading. Alternatively, OMMD was solubilized in water by addition of approximately 10% DMF and injected into the subphase of the Langmuir trough. The concentration of the spreading solutions varied from 0.2 to 0.34 mg/mL for the different polymers. The Langmuir layer was compressed and expanded by two barriers at a constant compression rate of 10 mm/min unless otherwise indicated. The surface pressure was recorded as a function of the mean molecular area per repeating unit (MMA). For OCL-OIBMD the repeating monomer unit (RPU) for determining MMA was calculated as sum of the molecular weights of ε-caprolactone, 3-(S)-iso-butyl-morpholine-2,5-dione, and 2,2,(4),4-trimethyl-hexamethylene-diisocyanate each multiplied with the respective percentage. For OIBMD and OSBMD the molecular weight of the repeating monomer unit of 171.2 g/mol and for OMMD the molecular weight of the repeating monomer unit of 129 g/mol was used to calculate the MMA. The experiments were performed at least three times. The surface potential (SP) was measured using a noncontact vibrating plate capacitor method. A computer-controlled device (KSV NIMA Surface Potential Sensor, SPOT, KSV Ltd, Finland) with a 16 mm diameter active electrode was placed at less than 3 mm above the air–water interface and a stainless steel reference electrode was immersed into the subphase.
The visualization of Langmuir monolayers was performed with the nanofilm_ep3 ellipsometer or a nanofilm_ultrabam (both Accurion GmbH, Goettingen, Germany) equipped with a 50 mW laser emitting p-polarized light at a wavelength of 658 nm, a 10× magnification objective, and a CCD camera. BAM images have an area of 500 × 400 µm2 and a spatial resolution of 2 µm. BAM experiments were performed during compression and expansion isotherm experiments of the films.
PM-IRRAS spectra were recorded with a dedicated spectrometer from KSV-NIMA. The photoelastic modulator (Hinds Instruments) was operated in a way that the retardation between p- and s-polarization was λ/2 at 2700 cm−1. The angle of incidence was fixed at 74°. Each spectrum was generated by averaging over 2500 measurements lasting 200 ms each.
The copolymer was degraded in the semidilute regime with π degr = 5 mN/m on a phosphate buffered saline (PBS) subphase. After π degr was reached, a solution of Pseudomonas cepacia lipase was injected so that the final concentration of lipase in the subphase was 0.01 mg/mL. The surface pressure was kept constant by compressing the film to compensate for the dissolution of small chain fragments. OIBMD was degraded similarly at π degr = 7 mN/m but on a Milli-Q water subphase without enzymes.
AFM
OIBMD monolayers were transferred to silicon wafer substrates via Langmuir–Blodgett transfer at different surface pressures. The silicon substrates were hydrophilized by cleaning with an air-plasma for 10 min. AFM measurements were carried out in tapping mode with a JPK nanowizard 4. OLYMPUS OMCL-AC cantilevers were used (nominal spring constant 26 N/m). Spectra were flattened and smoothed with WSxM.[Reference Horcas, Fernández, Gómez-Rodríguez, Colchero, Gómez-Herrero and Baro37]
Descriptive statistics and error consideration
The data presented in this study are both qualitative (AFM, PM-IRRAS, BAM, degradation curves, SP) and quantitative results. The main error of M n (GPC) arises from the calibration with standards. The deviation between a single standard's [η]M n and the calibration curve can be up to 10% which represents the maximum error of the measurement in universal calibration. OH-group titrations were carried out three times and blank values were determined two times. The main source of error was the unknown amount of water in the hygroscopic solvent DMF and the sample weighing error. The former introduced a variation in the blank and the latter a variation in the sample values. The error of M n (OH-group) was calculated as the weighted sum of both variations. Only one NMR measurement was carried out for each sample. The error for M n (NMR) was calculated via error propagation from the reliability of the 1H-NMR peak intensity. We adopted the reliability from a study of the peak intensity reliability in PMMA and solanesol[Reference Chûjô, Hatada, Kitamaru, Kitayama, Sato and Tanaka38] which was found to be ±3% for signals from protons that are abundant in the molecule (repeat units) and ±10% for protons that are scarce (end-groups).
The monolayer compression isotherms were repeated several times. Difference in collapse surface pressures was about 3%, difference in MMA was about 5%. Degradation experiments were conducted twice (OIBMD) or three times (OCL-OIBMD). Trends were similar but local fluctuations could be quite large. For OCL-OIBMD, the two curves with the highest agreement are shown. PM-IRRAS spectra were recorded twice and agreed qualitatively, but low signal to noise ratio and problematic background subtraction could cause shifts in apparent peak positions of approximately 10 cm−1. The spectrum with the higher signal to noise ratio is shown.
Results and discussion
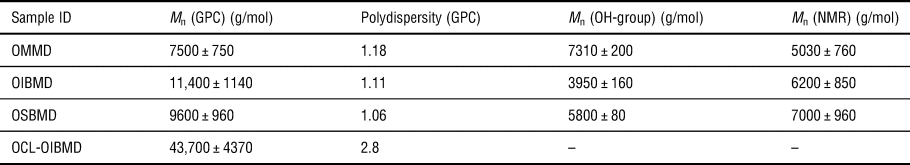
The molecular weights of the ODPs and the multiblock copolymer are listed in Table I. Analysis of the 1H-NMR spectrum (Supplementary Material S1) suggests that the multiblock copolymer contained roughly the same number of ε-caprolactone and depsipeptide repeat units (53:47).
Table I. Molecular weight of ODPs and one multiblock copolymer determined by multidetector GPC, OH-group titration, and 1H-NMR. The error considerations are explained in the Materials and methods section.
The π–A isotherms of OIBMD and OSBMD were recorded on a pure water subphase (pH 5.7) [Fig. 1(a)]. Changing the barrier speed from 10 to 40 cm2/min and the use of a PBS subphase (pH 7.4) had no significant influence on the shape and position of the isotherms (results not shown). The side-groups had a strong effect on the hydrophilicity of the depsipeptides. Although OMMD has comparable molecular weight to the other ODPs, it was almost impossible to dissolve OMMD in chloroform. Furthermore, a slight water-solubility was observed. Nevertheless, a mixture of chloroform and DMF (9.8/0.2 mL) allowed for spreading of the polymer at the air–water interface. An alternative pathway to the formation of OMMD monolayers is the preparation of a Gibbs adsorption layer by injection of an OMMD solution in water/DMF (DMF content of approximately 10% was required to solubilize OMMD in water) into the subphase. The final surface pressures in dependence on the subphase concentration are given in Fig. 1(c). A break in the logarithmic plot of surface pressure versus concentration is commonly identified with the critical micellar concentration (CMC). Following this argumentation, the CMC is about 0.2 µg/mL. These results suggest that OMMD could be applied as a biodegradable and potentially bioactive macromolecular surfactant.
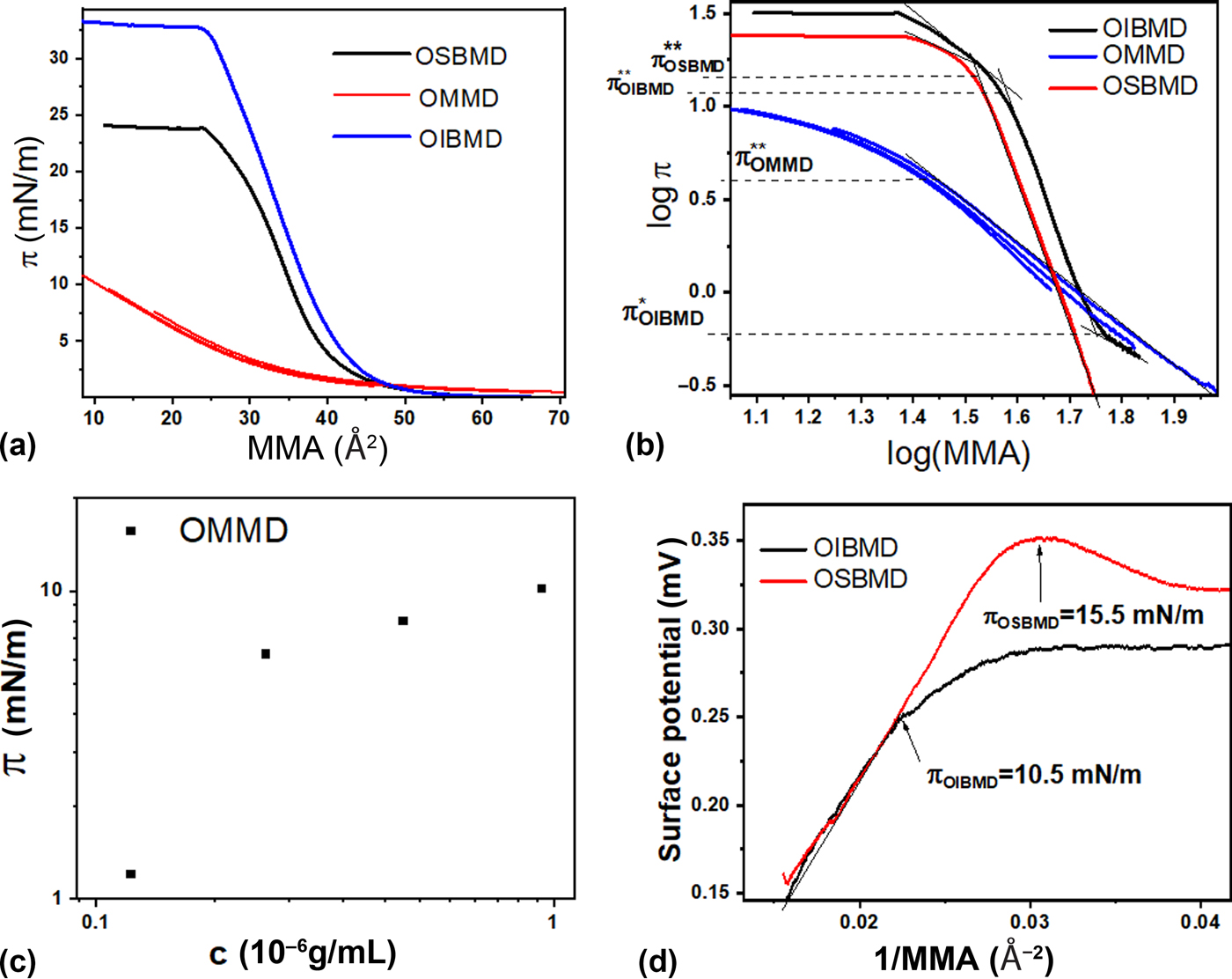
Figure 1. (a) Surface pressure versus area isotherms of the three ODPs. OMMD data is an overlay of three isotherms produced by spreading increasing volumes of polymer solution. (b) Logarithmic representation of the isotherms used to determine the 2D Flory exponent, the overlap concentration and the transition from semidilute to concentrated regime. (c) Logarithmic plot of surface pressure versus OMMD bulk concentration. (d) Plot of SP versus inverse of area per repeat unit (MMA) for OIBMD and OSBMD.
The shapes of the isotherms of OIBMD and OSBMD [Fig. 1(a)] are typical for macromolecules in relatively bad solvent conditions.[Reference Maestro, Ortega, Rubio, Rubio, Krägel and Miller39] A logarithmic plot of the isotherms [Fig. 1(b)] reveals the 2D Flory exponent v the semidilute regime according to 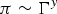 $\pi \sim {\rm \Gamma} ^y$ with y = 2v/(2v − 1) and
$\pi \sim {\rm \Gamma} ^y$ with y = 2v/(2v − 1) and 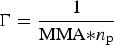 ${\rm \Gamma} = \displaystyle{1 \over {{\rm MMA*}n_{\rm p}}}$. The 2D Flory exponents are nearly identical for OIBMD (v = 0.58) and OSMD (v = 0.6), which is closer to the value expected for a theta solvent (v = 0.5) than for a good solvent (v = 0.75).
${\rm \Gamma} = \displaystyle{1 \over {{\rm MMA*}n_{\rm p}}}$. The 2D Flory exponents are nearly identical for OIBMD (v = 0.58) and OSMD (v = 0.6), which is closer to the value expected for a theta solvent (v = 0.5) than for a good solvent (v = 0.75).
The transition from semidilute to concentrated regime was observed at lower surface pressure for OIBMD (π** ≈ 12 mN/m) than for OSBMD (π** ≈ 16 mN/m) with OIBMD showing a much more extended concentrated regime (π collapse − π** ≈ 21 mN/m) than OSBMD (π collapse − π** ≈ 8 mN/m). Thus, the butyl side-group has a pronounced impact on the stability of the ODP monolayers. Consecutive compression/expansion cycles of OIBMD indicated that the chains behave differently in the concentrated state than in the semidilute state. Here, the isotherms were completely reversible when compression was halted in the semidilute state. When the films were compressed to the concentrated state, consecutive isotherms shifted to lower areas, indicating irreversible structure formation (Supplementary Material S2).
For OMMD layers, a very different behavior was observed. Here, the slope of the isotherm in a logarithmic plot is close to two, suggesting that ν is close to one [Fig. 1(b)]. This behavior is expected for the dilute state or a 2D gas of non-overlapping chains, indicating that the molecules avoid the formation of a semidilute state, e.g., by desorption. Since the air–water interface is such a good solvent for OMMD, the transition to the concentrated state occurred at very low area per repeat unit compared to OIBMD and OSBMD. Although the compression isotherm of OMMD did not show a horizontal plateau, our BAM observations (see below) indicate that OMMD underwent a “dynamic collapse” in the concentrated regime. With the length of a repeat unit being about 7.2 Å, it seems unlikely that a film can be compressed to an area of 10 Å2 per repeat unit and still be in the monolayer state. This hypothesis is supported by the observation of an exponential decrease of the surface pressure when the layer was first compressed to a surface pressure above π** and then held at constant surface area (Supplementary Material S3).
The SP of the OIBMD and OSBMD layers was analyzed to obtain further information about the orientation of the chains in the semidilute and concentrated state.
The surface pressure measures mainly the interaction between molecules in close contact (van der Waals interactions), whereas the SP measures the orientation of dipoles providing more long-ranged interactions. As expected, both OIBMD and OSBMD monolayers showed an increase of the SP as soon as the compression was started. According to the Helmholtz equation, the SP of a non-charged monolayer is 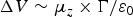 ${\rm \Delta} V \sim \mu _z \times {\rm \Gamma /} \varepsilon_0$ where Γ is the areal concentration and ε 0 the vacuum permittivity. Thus, when plotting ΔV over the inverse of the area per repeat unit [Fig. 1(d)], a linear relation indicates that the dipole moment of the molecules perpendicular to the water surface (μ z) does not change.
${\rm \Delta} V \sim \mu _z \times {\rm \Gamma /} \varepsilon_0$ where Γ is the areal concentration and ε 0 the vacuum permittivity. Thus, when plotting ΔV over the inverse of the area per repeat unit [Fig. 1(d)], a linear relation indicates that the dipole moment of the molecules perpendicular to the water surface (μ z) does not change.
For both OIBMD and OSBMD, a nearly linear relation was observed up to a surface pressure close to π**, indicating that the chains were reorienting when entering the concentrated regime. The initial slope was identical for both macromolecules, revealing identical orientation in the semidilute state. However, OSBMD could be compressed to a higher packing density and hence a higher SP before entering the concentrated regime. The reorientation of OSBMD in the concentrated regime was more pronounced than for OIBMD, leading to a reduction of the net dipole moment perpendicular to the air–water interface. In the collapsed state, the areal packing density cannot be further increased. Thus, the dipole moment remained constant while three-dimensional (3D) structures were formed.
To further intensify the analysis of the chain orientation, PM-IRRAS spectra were recorded [Fig. 2(a)]. The two-bands characteristic for the amide groups were observed at about 1630/cm (amide I) and 1530 to 1570 cm−1 (amide II). Furthermore, three bands characteristic of the ester groups were observed at 1725 cm−1 (C═O stretching), 1225 cm−1 (asymmetric COC stretching) and 1100 cm−1 (C―O stretching).[Reference Elzein, Nasser-Eddine, Delaite, Bistac and Dumas40] The selection rule of PM-IRRAS predicts that oscillators oriented preferentially parallel to the air–water interface produce an upward signal while oscillators oriented preferentially perpendicular to the air–water interface would produce a dip. Oscillators with random orientation generate no signal at all.[Reference Blaudez, Turlet, Dufourcq, Bard, Buffeteau and Desbat41] With our setup, we have yet to observe a credible dip, so we will not make assumptions about perpendicular orientations. No stretching bands of the methylene groups near 3000 cm−1 were observed at any surface pressure and the C═O stretching band of the ester group was much weaker than the amide bands. This strongly suggests that the ester groups were randomly oriented, meaning that these groups were not participating in hydrogen bonding and that there was no crystallization in the collapsed state. The asymmetric COC oscillators were oriented along the chain axis while the C―O oscillators were oriented in the carbonyl plane of the ester groups with a perpendicular and a parallel component with respect to the chain axis.[Reference Elzein, Nasser-Eddine, Delaite, Bistac and Dumas40] Thus, the relatively high intensities of the former two vibrations suggest that the chains were oriented predominantly parallel to the air–water interface at the ester group. The low intensity of the C═O stretching band in the semidilute state suggests that plane of the carbonyl bonds was rotating. In the concentrated and collapsed state (π = 20 mN/m to π = 32 mN/m), an increase of the C═O stretching band indicates that the rotation of the ester groups was reduced as the carbonyl planes became more parallel to the air–water interface. These observations are in agreement with the intensities of the amide vibrations. In the semidilute regime, amide II was not observed while amide I was relatively strong. This suggests that the amide planes were tilted so that the chain adopted a zigzag conformation. [Fig. 2(b)]. The low frequency of amide I indicates strong hydrogen bonding.[Reference Goormaghtigh, Cabiaux and Ruysschaert42] Thus, the conformation was similar to a beta sheet, but with every second amide group replaced by an ester group. Here, the chain is free to rotate, because these groups are not held in place by strong hydrogen bonds. It was also predicted by simulations that the additional flexibility introduced by the ester groups leads to novel secondary structures in depsipeptides compared to peptides, and this somewhat disordered pleated sheet might be one of them.[Reference Zhang, King, Suggs and Ren43] Interestingly, the main amide I band in electrospun samples of OCL-OIBMD block copolymers was at a higher frequency which agreed more with an alpha helix or, more probably, random coil conformation.[Reference Feng, Liu, Ren, Lu, Guo, Behl, Lendlein and Zhang31] This might be due to the fact that the molecules had little time to organize during electrospinning.
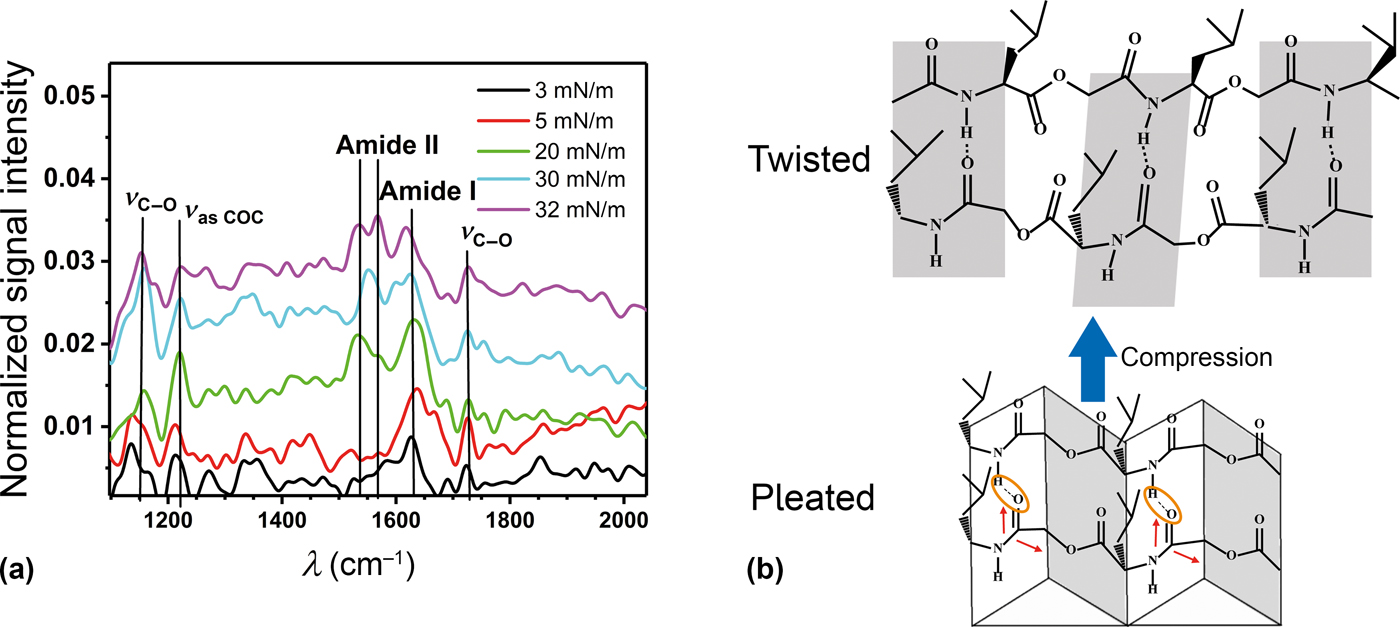
Figure 2. (a) PM-IRRAS spectra of OIBMD at different surface pressures. Normalized signal intensity is calculated by dividing the sample spectrum from the bare water spectrum and subtracting 1. (b) Sketch of the organization of the OIBMD chains. In the dilute state, the PM-IRRAS spectra indicate a pleated organization similar to a β-sheet while in the concentrated regime, an organization with twisted hydrogen bonding planes is suggested. The red arrows indicate the orientation of the transition dipole moments in the amide plane.[Reference Itoh and Oguri52]
In the concentrated regime, we observed a strong increase of amide II compared to amide I. This indicates that the zigzag of the amide planes was reduced, while the C═O bonds became more parallel to the surface normal. However, the constant frequency of amide I shows that hydrogen bonding was not substantially weakened. These observations can be explained when the chains were oriented parallel or antiparallel while the hydrogen bonding planes were twisted around the long axis [Fig. 2(b)].
Comparing these observations to the results from the SP measurements suggests that the SP in the z-direction was generated by the zig-zag of the chains. The weakening of the zigzag conformation in the concentrated state therefore explains the decrease of the dipole moment in the z-direction.
BAM was used to further analyze the film morphology. As expected, the layers were homogeneous in the semi-dilute state. In case of OIBMD (Fig. 3, middle) and OSBMD (Fig. 3, top), structure formation was observed immediately after entering the collapse regime indicated by a horizontal plateau of the isotherm. As described above, for OMMD, no horizontal plateau was observed, yet the film still collapsed according BAM images (Fig. 3, bottom). For all depsipeptides studied here, collapse occurred by formation of irregular and thicker structures at high areal density. Upon relaxation, the areal density of these 3D structures decreased while their size increased, indicating a reorganization driven by minimization of the surface energy of the collapsed layer. To investigate the structure formation of the layers at the nanometer scale, OIBMD films were transferred to silicon substrates with the Langmuir–Blodgett technique. The only distinguishable structural motifs were pebble-like structures (Fig. 4). The diameter of these pebbles decreased from about 50 to 20 nm when the film was compressed from the semidilute to the concentrated regime. The roughness of all films was very low, on the order of 2 Å, which also reflects the height of the pebbles. Their lateral dimensions agree reasonably well with the dimensions expected for the OIBMD chains in a “flattened coil” or “pancake” conformation, with 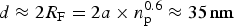 $d\approx 2R_{\rm F} = 2a \times n_{\rm p}^{0.6} \approx {\rm 35}\,{\rm nm}$ where we assumed that the length of the monomers a is 7.2 Å and n p is the degree of polymerization. Thus, in contrast to polypeptides like poly(γ-benzyl-glutamate), depsipeptides like OIBMD are not forming supramolecular assemblies. The beta-sheet like organization did not persist over long distance. The observation of no long-range order in ultrathin films of OIBMD is also in agreement with spin-coated films of OCL-OIBMD,[Reference Yan, Fang, Weigel, Behl, Kratz and Lendlein44] where crystallization was only observed after mobility was increased by thermal annealing of the films. Thus, the mobility of OIBMD in thin films at room temperature is too low for the formation of ordered structures.
$d\approx 2R_{\rm F} = 2a \times n_{\rm p}^{0.6} \approx {\rm 35}\,{\rm nm}$ where we assumed that the length of the monomers a is 7.2 Å and n p is the degree of polymerization. Thus, in contrast to polypeptides like poly(γ-benzyl-glutamate), depsipeptides like OIBMD are not forming supramolecular assemblies. The beta-sheet like organization did not persist over long distance. The observation of no long-range order in ultrathin films of OIBMD is also in agreement with spin-coated films of OCL-OIBMD,[Reference Yan, Fang, Weigel, Behl, Kratz and Lendlein44] where crystallization was only observed after mobility was increased by thermal annealing of the films. Thus, the mobility of OIBMD in thin films at room temperature is too low for the formation of ordered structures.
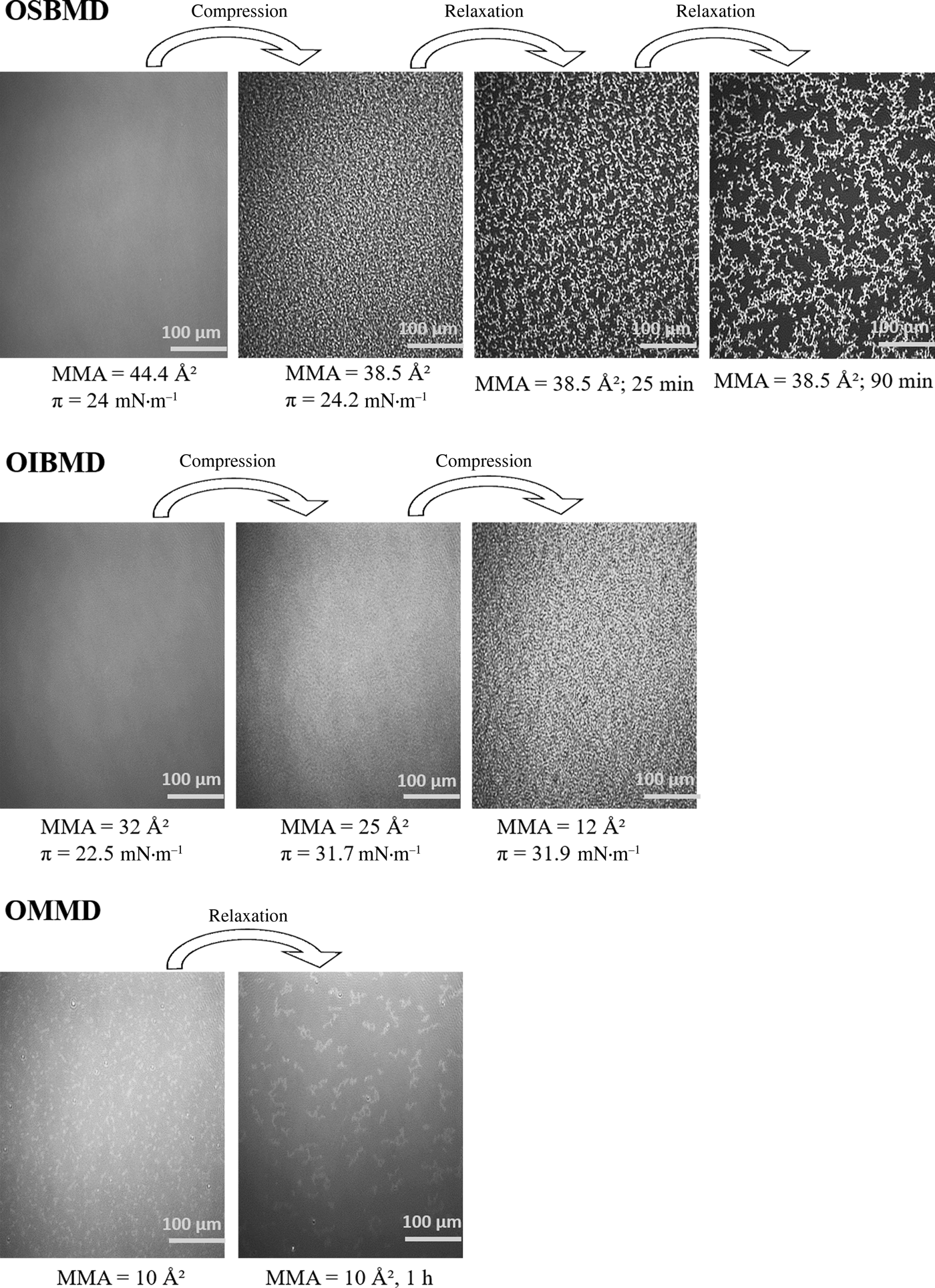
Figure 3. BAM images of OSBMD (top), OIBMD (middle), and OMMD (bottom).

Figure 4. AFM-height images of OIBMD films transferred to silicon substrates at different surface pressures. The height scale is identical for all images.
For medical applications where resorbable materials with shape-memory capabilities are required, multiblock copolymers of OCL and OIBMD are highly promising candidates. Here, we evaluated the impact of joining both oligomers with urethane linkers on the interfacial activity, molecular organization, and molecular degradation kinetics.
At the air–water interface, the 2D Flory exponent of the copolymer ν = 0.65 was higher than for pure OIBMD but lower than for pure OCL (ν = 0.77)[Reference Li and Esker45] [Fig. 5(b)]. The kink in the isotherm [Fig. 5(a)] was at nearly identical position (π = 13 mN/m, MMA = 21 Å2) as the kink in the isotherm of low molecular weight OCL (π = 12 mN/m, MMA = 21 Å2).[Reference Li and Esker45]
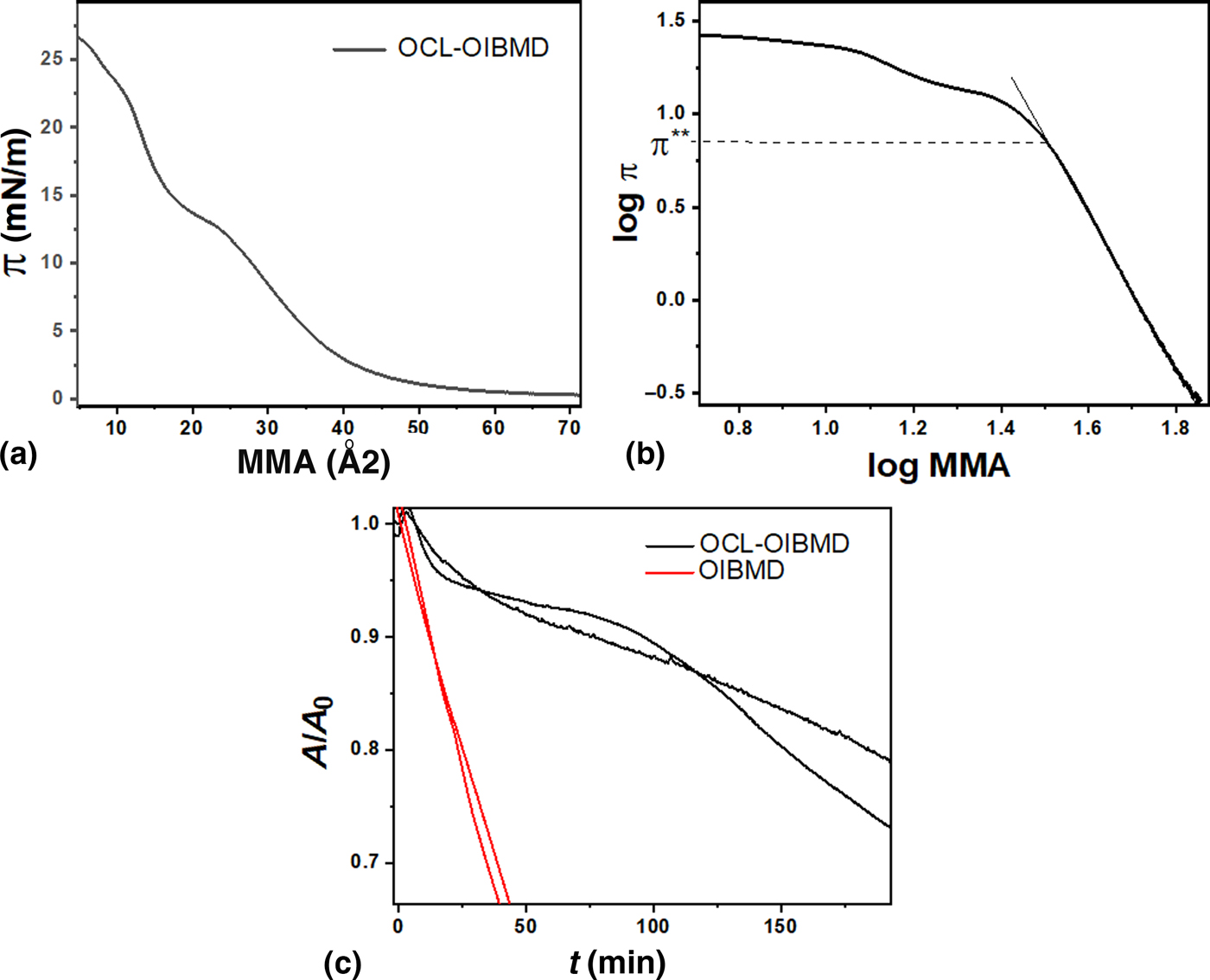
Figure 5. (a) Surface pressure versus area plot of OCL-OIBMD. (b) Logarithmic representation of the isotherms used to determine the 2D Flory exponent and the transition from semidilute to concentrated regime. (c) Isobaric degradation curves of OCL-OIBMD (black) and OIBMD (red). Both macromolecules were degraded at similar surface pressures in the semidilute state. Experiments were carried out twice for OIBMD and three times for OCL-OIBMD because variation was higher for OCL-OIBMD than for OIBMD. The two curves with the greater agreement are shown for OCL-OIBMD.
For pure OCL, the kink was followed by a depression of the isotherm upon further compression, which was accompanied by formation of spherulitic crystals visible in BAM. In contrast, the OCL-OIBMD block copolymer showed no depression and no spherulitic crystals were visible in BAM. These observations suggest that the crystallization of the OCL segments is hindered severely by the presence of the OIBMD blocks. We note that this was also observed in the bulk materials, where OCL crystallization could only be observed after stretching,[Reference Yan46] and especially in thin spin coated OCL-OIBMD films, where OIBMD crystallization was only observed after annealing, and PCL crystallization was severely hindered.[Reference Yan, Fang, Weigel, Behl, Kratz and Lendlein44] This was attributed to the reduced mobility of OCL blocks anchored to OIBMD chains, which are glassy at room temperature. However, since the air–water interface is a reasonably good solvent for OCL-OIBMD, dynamics should be much faster than in thin films. Previous examinations of block copolymers at the air–water interface strongly suggest that these polymers form phase-separated Langmuir films. Since the microdomains are very small, probably containing only few chains,[Reference Aoki, Kunai, Ito, Yamada and Matsushige47] crystallization inside these domains is either impossible or leads to nanocrystals that are not observable in BAM. The reversibility of the isotherms was identical to the OIBMD homopolymer: when compressed to the concentrated state, consecutive isotherms shifted to lower area.
Isobaric, enzymatic degradation curves of OCL-OIBMD and isobaric hydrolytic degradation curves of OIBMD are shown in Fig. 5(c). The enzymes can only degrade the OCL blocks in the copolymer. There are currently no hydrolytic bulk degradation experiments of OIBMD molecules in the literature, but according to degradation experiments with OCL-OIBMD multiblock copolymers,[Reference Feng, Behl, Kelch and Lendlein24] it is expected that the oligo-depsipeptides degrade via fast hydrolysis of the ester bonds between amino acid and glycine. This hypothesis was confirmed by our Langmuir monolayer degradation experiments. The degradation rate of OCL-OIBMD was much lower than the degradation rate of both pure OIBMD as well as the degradation rate of pure OCL at similar concentrations of enzymes.[Reference Schöne, Kratz, Schulz and Lendlein48] It is therefore clear that the presence of OIBMD blocks hinders the enzymatic degradation of OCL segments. The retardation of the enzymatic degradation of OCL blocks by enzymatically non-degradable blocks has been observed before.[Reference Schöne, Kratz, Schulz and Lendlein49] Concerning the mechanism, the degradation of OCL-OIBMD showed the typical convex curve shape of a random fragmentation mechanism. The initially low dissolution rate is characteristic for block copolymers and high molecular weight polymers. Small, water soluble fragments can be generated either by single chain cuts or several consecutive chain-cuts. Naturally, fragment generation via several consecutive chain-cuts takes longer than by single chain-cuts. Since only single cuts near the chain ends lead to small fragments, the higher the molecular weight, the more delayed the degradation. In the case of multiblock copolymers containing non-degradable blocks and linkers, many chain-ends may be non-degradable, which reduces the initial degradation rate even further. For longer degradation times (150 min), the degradation rate of OCL-OIBMD became comparable to the degradation rate of OIBMD. A random fragmentation mechanism was also supported by bulk degradation experiments of OCL-OIBMD,[Reference Brunacci, Wischke, Naolou, Neffe and Lendlein27] where a fast decrease of the molecular weight at low mass loss was observed. The fast decrease of the molecular weight is caused by the rapid fragmentation of OIBMD segments, while the slow diffusion of fragments through the bulk material limits mass loss to the surrounding medium. In bulk OCL-OIBMD, the diffusion of OIBMD fragments is further hampered by the slowly degrading OCL domains.
Conclusion
The behavior of morpholine-2,5-dione-based ODPs at the air–water interface was investigated using the Langmuir technique. The solvent quality of the air–water interface depended on the length of the hydrophobic side-group. The butyl derivatives were in rather bad solvent conditions while the slightly water-soluble methyl variant was under good solvent conditions. The collapse surface pressure of the sec-butyl variant was higher than for the isobutyl variant, which allowed these molecules to achieve slightly higher packing density. While the collapsed films showed evolving structures at the micrometer level, PM-IRRAS measurements showed that these aggregates were non-crystalline. This is a marked difference to the situation in bulk, where all of the investigated oligomers and the multiblock copolymer were able to crystallize. On the other hand, the crystallization of PCL, which is usually observed in Langmuir films, was suppressed in OCL-OIBMD block copolymer monolayers. The hindering of OCL crystallization in OCL-OIBMD block copolymers in bulk is attributed to the slow dynamics of the OIBMD microdomains, which are crystalline or glassy below the melting temperature of PCL. In Langmuir films, we suspect that the reason for the impeded crystallization is rather the small number of chains in the microdomains than slow dynamics, but this requires further characterization.
PM-IRRAS spectroscopy of OIBMD revealed a secondary structure reminiscent of a beta sheet, with strong hydrogen bonds between amide groups but little preferential orientation at the ester bonds. To our knowledge, this represents the first attempt to observe the organization of ODPs at the molecular level. The hypothesis of a beta-sheet like organization is in agreement with x-ray scattering of bulk OIBMD,[Reference Yan, Fang, Noechel, Gould, Behl, Kratz and Lendlein50] where the strongest reflection was at a d-spacing of about 10 Å, which agrees very well with the d-spacing in the fiber direction of antiparallel beta sheet crystals.[Reference Kajava, Squire and Parry51] According to AFM images of transferred films, the weaker intramolecular interactions compared to peptides seem to hinder the formation of long-range order or supramolecular structures. The lack of supramolecular organization is unexpected because the amino acids alanine (OMMD), leucine (OIBMD), and isoleucine (OSBMD) are known to stabilize helical conformations and helices have a tendency to form fibrils. Polydepsipeptides that assume helical conformations could show liquid crystalline behavior, which would be very interesting for a great number of applications. To further promote helix formation, side-chains could be introduced to the α-hydroxy acids, or the amino acids could be replaced by γ-benzylglutamic acid.
The depsipeptides showed fast hydrolytic degradation both alone and when used as building blocks for multiblock copolymers, which is in qualitative agreement with the expectations based on bulk degradation experiments. The more detailed understanding of the impact of the unique molecular architecture of ODP-based building blocks on their interfacial behavior will enable the development of the next biomaterial generation with tailored properties for highly sophisticated application in gene delivery or tissue engineering.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1557/mrc.2019.21.
Acknowledgments
The authors thank Dr. Marc Behl for providing the oligomers and the multiblock copolymer. This work was supported by the Helmholtz-Association through program-oriented funding.










 Open access
Open access