



Introduction
In our Core Facility, we are presented with a wide variety of tissue and sample types to process for TEM observation. Some tissues, such as liver, do not present orientation problems. Other tissues such as skeletal muscle have specific orientations that must be maintained. As long as the initial dissection of the tissue is done so that the orientation or area of interest can be identified, these samples do not present a problem. Likewise, when it is not necessary to maintain orientation, cells in culture can be treated with trypsin, to release them from the plate, and pelleted. Or the monolayer can be fixed in vitro and then scraped and pelleted, and the pellet can be processed while adherent to the wall of the Eppendorf tube or handled like a small piece of tissue, among other methods.
When it is imperative that the orientation of the monolayer be maintained, as with a polarized epithelial cell type or multiple cell types in co-culture, the cells must remain in place throughout processing and embedding. Cells can be grown on coverslips, fixed and processed, and then inverted over filled embedding capsules, but then the glass must be removed. Methods for doing this include etching the glass away with HF acid—a highly dangerous substance best avoided if possible. Or the coverslip can be “popped” off by heating the block and then rapidly cooling it in liquid nitrogen. In my hands, this latter method has always been hit-or-miss.
Over the years I have come to use two methods for handling oriented cell monolayers that have proven to be quite reliable. I am not claiming that these are unique to me. In fact, I owe the second method to Thierry Gilbert, who was a post-doc in cell biology at Weill Cornell Medical College in the late 1980s. I am just passing it along. We all learn from one another.
Method 1
Adherent cells are grown in a polystyrene culture dish. This can be a single dish, a multi-well plate (6 wells work well), or even a t-25 flask as shown (Figure 1). If flasks are used, the top surface of the flask is removed by cutting it with a fine-tipped soldering iron during one of the buffer rinse steps (Figure 2), being careful not to breath the fumes generated; they are toxic. The cells are fixed, post-fixed, en bloc stained with uranyl acetate, and then dehydrated through a graded ethanol series up to 3 changes in 100%. Following the last change of 100% ethanol, the alcohol is drained as completely as possible by inverting the dish over paper toweling, and the cells are immediately overlaid with a thin layer of embedding resin, using LX-112 as the epoxy component. This is allowed to sit for 30 minutes to allow extraction of the last of the ethanol. The resin is removed and replaced with a thin layer (2 mm) of fresh resin, and embedding tubes (made by cutting the pyramidal ends from BEEM capsules) are inserted into the resin, with the even, machined top of the tube down into the resin. If there are areas of interest, they can be targeted at this time.

Figure 1: Cell monolayers in T-25 flasks.

Figure 2: Cutting the lid from the flask.
The dish is placed in the oven overnight to begin the polymerization. The next day, labels are inserted into the tubes, and the tubes are filled to the top with fresh resin (Figure 3). The whole assemblage is returned to the oven to complete polymerization. The blocks are separated from the dish by grabbing the tube with needle-nosed pliers and snapping it away with a shearing motion (Figure 4). The blocks will separate fairly cleanly. Some will take along a bit of the culture dish's plastic, but this can be easily removed when the block is being trimmed for sectioning (Figure 5).

Figure 3: Embedding tubes filled with resin on the second day.

Figure 4: Removing polymerized blocks by snapping with shearing force.

Figure 5: Raw block face (top). Block face with dish remnant peeled away (middle). Trimmed block face (bottom).
Sections are cut en face. Initial semi-thin sections are cut at 200 nm, placed on a slide, and stained with Toluidine Blue (1% in 1% sodium borate) for evaluation. Ultrathin sections are then cut and collected, contrasted, and viewed as usual (Figure 6).
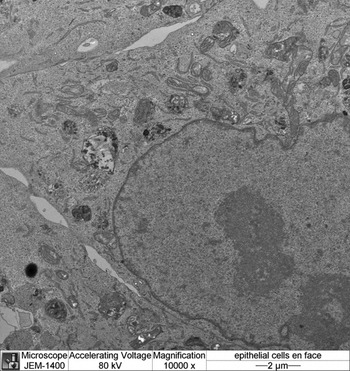
Figure 6: En face image of a urinary bladder epithelial cell monolayer.
Note on materials: After experimenting with many permutations of resin components, I found that it is imperative to use the LX-112 epoxy resin (Ladd Research Industries, Burlington, VT). All other components can be from your vendor of choice. I use those from Electron Microscopy Sciences (Hatfield, PA): LX112 9.7 gm, DDSA 3.2 gm, NMA 0.9 gm, and BDMA 34 microliter/ml of total volume.
Method 2
This method allows for examination of the cells in cross section, rather than en face. This can be invaluable when studying polarized epithelia or other cell types with specific basal-to-apical distribution of organelles.
Cells are grown to confluence on polycarbonate (opaque) Costar Transwell filters (Corning Life Sciences). The culture is rinsed with serum-free medium, fixed, and post-fixed as usual. The membrane is cut free of its holder (Figure 7), and the sample is dehydrated through a graded ethanol series. After the last ethanol, the filter is placed in fresh, anhydrous propylene oxide and gently shaken. The filter will roll onto itself, forming a “jelly roll.” This roll is then placed in an Epon-analog embedding resin for 30 minutes to allow infiltration of the resin. The roll can be cut into shorter lengths and then embedded in a flat mold (Figure 8). When this block is trimmed and cut, one-micrometer sections reveal cells on the rolled filter (Figure 9). The roll is often tight enough that it can be contained in its entirety in the block face, providing a large number of cells for study (Figure 10).

Figure 7: Polycarbonate filter separated from its holder after osmium post-fixation.

Figure 8: Polycarbonate filter curled in propylene oxide (top). Curled filter cut into pieces and embedded (bottom).

Figure 9: One-micrometer-thick section of rolled filter. Toluidine Blue staining.

Figure 10: MDCK cells on polycarbonate filter. Note the tight-junction complex extending from the apical membrane.
Acknowledgments
My thanks to Drs. Diane Felsen and Geri Kreitzer for providing the cell cultures used in the preparation of this manuscript, and to Rebecca Smith for her help with the sample preps and for reading the manuscript.










