1 results
Synthesis and structural characterization of 3-[1-[4-(2-methylpropyl)phenyl]ethyl]-6-(4-fluorophenyl)-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazole
-
- Journal:
- Powder Diffraction / Volume 34 / Issue 4 / December 2019
- Published online by Cambridge University Press:
- 02 September 2019, pp. 325-330
-
- Article
- Export citation
-
3-[1-[4-(2-Methylpropyl)phenyl]ethyl]-6-(4-fluorophenyl)-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazole (C21H21FN4S) has been synthesized as a member of a series of triazolothiadiazoles having NSAIDs moieties with cytotoxic activity. The crystal structure of this new compound has been solved and refined using conventional laboratory X-ray powder diffraction data and optimized using density functional techniques. The final structure solution was achieved by Rietveld refinement using soft restraints on all non-H atom bond lengths and angles. This compound crystallizes in
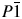 $P\bar{1}\;$ space group, with the unit cell parameters a = 5.5880(4) Å, b = 9.3074(7) Å, c = 19.497(4) Å, α = 99.311(10)°, β = 91.925(9)°, γ = 98.199(6)°, and V = 988.8(2) Å3. To complement and verify the structure solution of the compound, the density functional theory (DFT) calculations were performed by using the local density approximation and the generalized gradient approximation for exchange-correlation energy. In order to see the effect of the van der Waals interactions on the electronic structure, the relevant structure was also optimized with B3LYP-D2, PBE-D2, and optB88-vdW functionals. The refined crystal structure was confirmed by the DFT calculations. The best agreement with the experimental structure was achieved by optB88-vdW functional.
$P\bar{1}\;$ space group, with the unit cell parameters a = 5.5880(4) Å, b = 9.3074(7) Å, c = 19.497(4) Å, α = 99.311(10)°, β = 91.925(9)°, γ = 98.199(6)°, and V = 988.8(2) Å3. To complement and verify the structure solution of the compound, the density functional theory (DFT) calculations were performed by using the local density approximation and the generalized gradient approximation for exchange-correlation energy. In order to see the effect of the van der Waals interactions on the electronic structure, the relevant structure was also optimized with B3LYP-D2, PBE-D2, and optB88-vdW functionals. The refined crystal structure was confirmed by the DFT calculations. The best agreement with the experimental structure was achieved by optB88-vdW functional.