



Impact statement
Leukaemia describes a mixed group of cancers affecting blood cell development. Its management has changed drastically over the past 40 years and with this, we have seen significant improvements in patient outcomes. These advances have come about through continued research into the underpinning mechanisms driving leukaemia development. In addition to better treatments for leukaemia, our understanding of the genes that cause leukaemia has improved. This review focuses on the blood cancer-causing gene BCR::ABL1 which results from an abnormal join between two chromosomes, 9 and 22. The BCR::ABL1 gene most commonly leads to a blood cancer called chronic myeloid leukaemia (CML), but can also cause an acute leukaemia, more commonly acute lymphoblastic leukaemia and rarely, acute myeloid leukaemia. Our knowledge of how the BCR::ABL1 gene and resulting protein drive cancer cell progression and survival through complex processes within the cell which promote cell growth and switch off normal cell death pathways. At the same time, targeted treatments have been developed which directly stop BCR::ABL1 from affecting cell growth and survival, replacing traditional, more toxic chemotherapy drugs which have a blanket effect on all developing cells, both healthy and cancer cells. These new drugs are called tyrosine kinase inhibitors or “TKIs”; the first of which was designed nearly 25 years ago and is now called imatinib (trade name “Gleevec” or “Glivec”). TKIs have led to much improved survival for patients, especially with CML, but also reduced toxicity and improved quality of life. Indeed, patients with the most benign phase of CML, termed chronic phase, if responding to these targeted drugs, can expect normal life expectancy. This paper will discuss the advances made in BCR::ABL1-positive leukaemias, and discuss ongoing issues with their use and highlight where research must now focus to continue to improve outcomes for patients through precision medicine.
Background
First described in the 19th century, the term “leukaemia” describes a heterogenous group of conditions affecting the various levels of haematopoietic cell development (Bennett, Reference Bennett1845). More recently, our understanding of leukaemias continues to develop, and with advances in genetic and epigenetic mechanisms, as well as diagnostics, our appreciation of the underpinning pathogenesis leading to these malignant clones has brought with it targeted treatment advances.
Chronic myeloid leukaemia (CML) represents an excellent model where understanding of disease pathogenesis has been translated into clinical practice, and continues to improve outcomes for patients. This article will focus on the development of tyrosine kinase inhibitors (TKIs) in CML, as well as the emerging specifically targeting the ABL myristoyl pocket (STAMP) inhibitors. We will go on to discuss the side-effect profiles relating to both “on-target” and “off-target” mechanisms via kinase inhibition, and briefly discuss their emerging roles in Philadelphia chromosome-positive acute lymphoblastic leukaemia (Ph+ ALL) and Ph+ acute myeloid leukaemia (Ph+ AML).
Chronic myeloid leukaemia and the discovery of BCR::ABL1
CML accounts for 15–20% of all adult leukaemias with an incidence of around 1 in 100,000. Median presentation is between 40 and 60 years. Classically, patients present in chronic phase (CP) disease with high white cell count and splenomegaly, and without medical intervention, will progress to blast phase (BP), which behaves like an acute leukaemia with dismal outcomes. CML was previously described as a triphasic disease, but recent WHO guidelines have removed accelerated phase as a disease entity, in keeping with recent scientific advancement (Khoury et al., Reference Khoury, Solary, Abla, Akkari, Alaggio, Apperley, Bejar, Berti, Busque, Chan, Chen, Chen, Chng, Choi, Colmenero, Coupland, Cross, de Jong, Elghetany, Takahashi, Emile, Ferry, Fogelstrand, Fontenay, Germing, Gujral, Haferlach, Harrison, Hodge, Hu, Jansen, Kanagal-Shamanna, Kantarjian, Kratz, Li, Lim, Loeb, Loghavi, Marcogliese, Meshinchi, Michaels, Naresh, Natkunam, Nejati, Ott, Padron, Patel, Patkar, Picarsic, Platzbecker, Roberts, Schuh, Sewell, Siebert, Tembhare, Tyner, Verstovsek, Wang, Wood, Xiao, Yeung and Hochhaus2022).
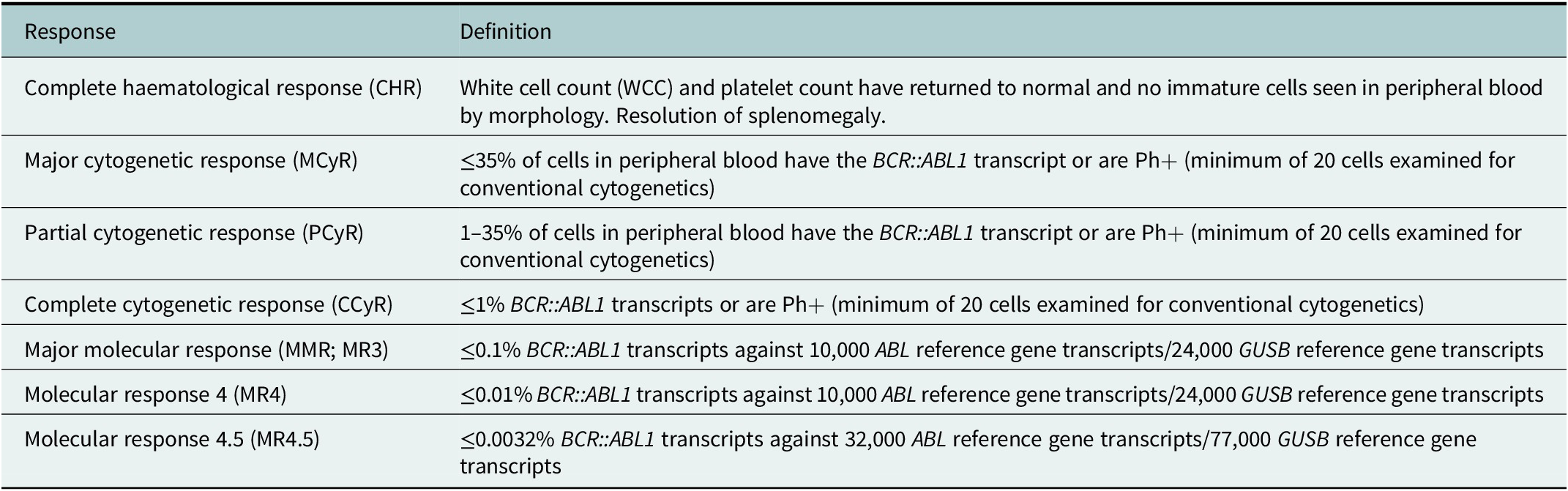
Historically, initial treatment involved standard cytotoxic chemotherapy, including busulphan and hydroxycarbamide. Definitions of treatment response in CML are described in Table 1. Although a haematological response was obtained with this approach, no cytogenetic response could be achieved as BCR::ABL1 was not being targeted specifically (Hehlmann et al., Reference Hehlmann, Heimpel, Hasford, Kolb, Pralle, Hossfeld, Queisser, Löf-fler, Heinze and Georgii1993). Interferon alpha was introduced as a standard-of-care in the mid-1980s, and demonstrated a significant improvement when compared to preceding therapies, with a 15% higher 5-year survival (Talpaz et al., Reference Talpaz, Kantarjian, McCredie, Trujillo, Keating and Gutterman1986). Cytogenetic response and overall survival were improved when combined with systemic chemotherapy, such as cytarabine (Beck et al., Reference Beck, Guilhot, Giles, Aoki, Wirt and Guilhot2001). Within the 1980s, it also became clear that allogeneic haematopoietic stem cell transplantation (alloHSCT) could result in long-term disease-free survival and probably cure of the disease for selected patients, though not without significant risk of morbidity and mortality (Fefer et al., Reference Fefer, Cheever, Thomas, Boyd, Ramberg, Glucksberg, Buckner and Storb1979; Clift et al., Reference Clift, Buckner, Thomas, Doney, Fefer, Neiman, Singer, Sand-ers, Stewart, Sullivan, Deeg and Storb1982).
Table 1. Definitions of response in CML, international assessment scale
In 1970, Janet Rowley discovered the Philadelphia Chromosome in patients with CML, a shortened chromosome 22 as a result of a translocation with chromosome 9 (t(9;22)(q34;q11)) (Rowley, Reference Rowley1973). We now know this to result in the BCR::ABL1 fusion oncogene. The breakpoint in the Abelson (ABL1) gene is generally constant occurring after exon 2 (a2). In the breakpoint cluster region (BCR) gene, however, this is more variable, occurring most commonly after e13 or e14 in CML and e1 and e2 in Ph+ ALL and Ph+ AML. As a result, three chimeric BCR::ABL1 proteins of different molecular weight can be produced (p190, p210 and p230 BCR::ABL1) (Melo, Reference Melo1997). Rarely, observed variations in breakpoints have been observed (Arber et al., Reference Arber, Orazi, Hasserjian, Thiele, Borowitz, le Beau, Bloomfield, Cazzola and Vardiman2016). BCR::ABL1 encodes a constitutively active oncogenic tyrosine kinase leading to uncontrolled cell proliferation, antiapoptotic effects, arrested lymphoid development and multistep oncogenic progression.
The molecular hallmark expressed in the vast majority of CML cases is the p210 protein, also being expressed in around 25% of Ph+ ALL. The remaining 75% of Ph+ ALL express the p190 BCR::ABL1 protein, with only around 1–2% of CML cases expressing this variant. In addition, 5–7% of CML cases show co-expression of both p190 and p210 isoforms (Molica et al., Reference Molica, Zacheo, Diverio, Alimena and Breccia2015). The p190 protein lacks the DH–PH domain and when associated with CML is often associated with a monocytosis, absence of splenomegaly and bone marrow morphology which is intermediate between CML and chronic myelomonocytic leukaemia with inferior outcome and short-lived responses to TKIs (Gandhe et al., Reference Gandhe, Vekaria and Dabak2021). The difference between these two isoforms is still poorly characterised, although p190-specific up-regulation of IFN pathways and activation of STAT1 and STAT2 as well as Src and PAK1 kinases has been observed which may explain some of the differences in disease phenotype (Adnan-Awad et al., Reference Adnan-Awad, Kim, Hohtari, Javarappa, Brandstoetter, Mayer, Potdar, Heckman, Kytölä, Porkka, Doma, Sexl, Kankainen and Mustjoki2021). The p230 isoform is classically associated with neutrophilic CML (CML-N) demonstrating a lower total white blood cell count, less severe anaemia, less prominent splenomegaly and a more indolent course (Szuber and Tefferi, Reference Szuber and Tefferi2018).
Development of highly efficacious targeted therapeutics in the form of BCR::ABL1-specific TKIs has revolutionised the treatment of CML, leading to a more than 10-fold increase in survival for CML patients without the associated toxicities of alloHSCT. A number of scores including the SOKAL score and EUTOS long-term survival score (ELTS) have been developed to predict response to therapy in CML and are used in many of the studies we will discuss. However, work is still required to validate these scores in the current treatment landscape. ELTS has been demonstrated as a better predictor of outcome, however, further validation is required with the first-line use of second-generation TKI therapies (Pfirrmann et al., Reference Pfirrmann, Clark, Prejzner, Lauseker, Baccarani, Saussele, Guilhot, Heibl, Hehlmann, Faber, Turkina, Ossenkoppele, Höglund, Za-ritskey, Griskevicius, Olsson-Strömberg, Everaus, Koskenvesa, Labar, Sacha, Zackova, Vervantes, Colita, Zupan, Bogfanovic, Castagnetti, Guilhot, Hasford, Hochhaus and Hoffmann2020).
Monitoring response to therapy in CML
Monitoring of disease with the use of quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) is paramount to the effective use of TKIs for disease management. BCR::ABL1 transcripts are compared to a housekeeper gene (usually ABL1) allowing for very sensitive assessment of response to TKI therapy with detection of minimal residual disease using the international scale (Table 2), and decisions around switches in therapy to be made early (Hehlmann, Reference Hehlmann2020; Hochhaus et al., Reference Hochhaus, Baccarani, Silver, Schiffer, Apperley, Cervantes, Clark, Cortes, Deininger, Guilhot, Hjorth-Hansen, Hughes, Janssen, Kantarjian, Kim, Larson, Lipton, Mahon, Mayer, Nicolini, Niederwieser, Pane, Radich, Rea, Richter, Rosti, Rousselot, Saglio, SauBele, Soverini, Steegmann, Turkina, Zaritskey and Hehlmann2020; Smith et al., Reference Smith, Apperley, Milojkovic, Cross, Foroni, Byrne, Goringe, Rao, Khorashad, de Lavallade, Mead, Osborne, Plummer, Jones and Copland2020).
Table 2. Treatment milestones in CML

Note: BCR::ABL1 qRT-PCR results expressed on the international scale, highlighting optimal response, warning and treatment failure at specific time points.
First-generation TKIs
In 1996, preclinical data demonstrated that a modified 2-phenylaminopyrimidine induced apoptosis of BCR::ABL1-positive human cells, including primary CML cells, with little toxicity to normal cells (Druker et al., Reference Druker, Tamura, Buchdunger, Ohno, Segal, Fanning, Zimmermann and Lydon1996). This compound, now known as imatinib, specifically inhibited BCR::ABL1 tyrosine kinase activity by competitive inhibition at the ATP-binding site of the BCR::ABL1 protein. This resulted in the inhibition of phosphorylated proteins from downstream signalling pathways. Although imatinib effectively inhibits BCR::ABL1, it also blocks other kinases, including platelet-derived growth factor receptor (PDGFR) and c-KIT (Giles et al., Reference Giles, O’Dwyer and Swords2009). Its clinical ability was demonstrated in the International Randomised Study of Interferon and STI571 (IRIS) study, where imatinib (STI571) was shown to induce significantly higher rates of haematological and cytogenetic responses, and subsequently dramatically improved overall survival compared to the interferon-alpha plus low-dose cytarabine; the standard-of-care prior to TKIs (O’Brien et al., Reference O’Brien, Guilhot, Larson, Gathmann, Baccarani, Cervantes, Cornelissen, Fischer, Hochhaus, Hughes, Lechner, Nielsen, Rousselot, Re-iffers, Saglio, Shepherd, Simonsson, Gratwohl, Goldman, Kantarjian, Taylor, Verhoef, Bolton, Capdeville and Druker2003). In IRIS, initial assessment at a median of 19 months, demonstrated a complete cytogenetic response (CCyR) rate of 74% versus 9% (p < 0.001) and failure to progress at 12 months of 99% versus 93% (p < 0.001) in the imatinib arm compared to interferon-alpha plus low-dose cytarabine, respectively (O’Brien et al., Reference O’Brien, Guilhot, Larson, Gathmann, Baccarani, Cervantes, Cornelissen, Fischer, Hochhaus, Hughes, Lechner, Nielsen, Rousselot, Re-iffers, Saglio, Shepherd, Simonsson, Gratwohl, Goldman, Kantarjian, Taylor, Verhoef, Bolton, Capdeville and Druker2003).
Importantly, this response was durable, with an 8-year follow-up demonstrating event-free survival of 91% and overall survival of 93% (Hochhaus et al., Reference Hochhaus, O’Brien, Guilhot, Druker, Branford, Foroni, Goldman, Müller, Radich, Rudoltz, Mone, Gathmann, Hughes, Larson and IRIS2009). However, an 8-year follow-up within the trial highlighted that only 55% of patients enrolled remained on imatinib therapy, highlighting potential issues with tolerability and adverse events amongst this patient group (Table 3). In addition, 16% of patients discontinued treatment due to treatment resistance. Although dose escalation of imatinib can improve response rates in patients with a sub-optimal response, switching to second or third-generation TKIs can be more effective (Cortes et al., Reference Cortes, Jones, O’Brien, Jabbour, Konopleva, Ferrajoli, Kadia, Borthakur, Stigliano, Shan and Kantargian2010a, Reference Cortes, Jones, O’Brien, Jabbour, Konopleva, Ferrajoli, Kadia, Borthakur, Stigliano, Shan and Kantargianb; Kantarjian et al., Reference Kantarjian, Shah, Hochhaus, Cortes, Shah, Ayala, Moiraghi, Shen, Mayer, Pasquini, Nakamae, Huguet, Boqué, Chuah, Bleickardt, Bradley-Garelik, Zhu, Szatrowski, Shapiro and Baccarani2010). Since the introduction of imatinib, second and third-generation TKIs have been developed.
Table 3. Common side effects associated with individual TKIs, degree of severity typically related to effect on other kinase activity affected although in many cases full mechanism of side effects poorly understood
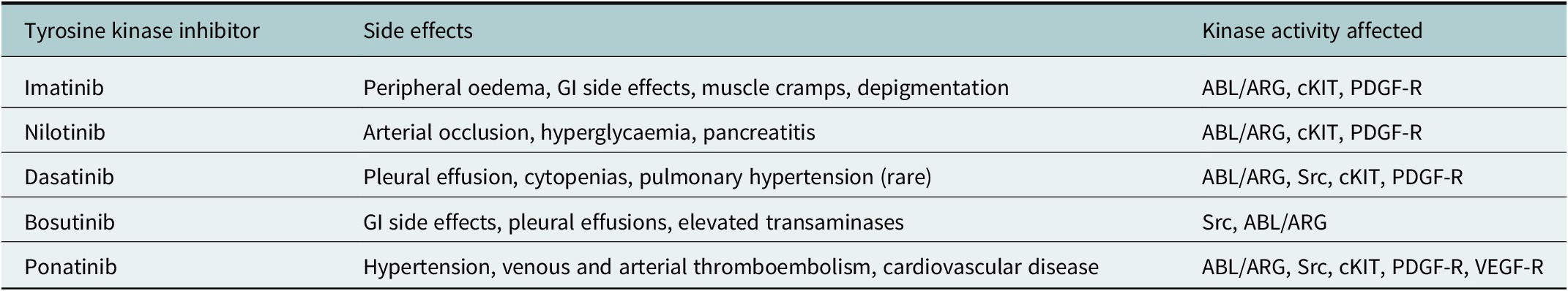
Note: Under physiological circumstances, ABL/ARG regulates response to DNA damage and oxidative stress; Src kinases involved with haematopoiesis, innate and adaptive immune response and vascular permeability; c-KIT regulates haematopoiesis, GI motility and melanogenesis; PDGF-R regulation of interstitial fluid pressures, VEGF-R cardiac homeostasis and angiogenesis.
Second- and third-generation TKIs
Dasatinib, nilotinib, bosutinib and ponatinib have led to further improvement in CML outcome. Dasatinib is a dual SRC/ABL1 second-generation TKI shown to be significantly more potent when compared to imatinib in vitro. The DASISION trial (NCT00481247) compared dasatinib 100 mg OD and imatinib 400 mg OD in the setting of front-line treatment (Kantarjian et al., Reference Kantarjian, Shah, Hochhaus, Cortes, Shah, Ayala, Moiraghi, Shen, Mayer, Pasquini, Nakamae, Huguet, Boqué, Chuah, Bleickardt, Bradley-Garelik, Zhu, Szatrowski, Shapiro and Baccarani2010). At 12 months, the primary outcome of confirmed CCyR was 83% versus 72% (p = 0.007) in the dasatinib and imatinib arms, respectively (Jabbour and Kantarjian, Reference Jabbour and Kantarjian2014). Five-year follow-up demonstrated that dasatinib achieved deeper responses at earlier time points, with a higher proportion of patients achieving major molecular response (MMR) at 3 months in the dasatinib arm (84% versus 64%, p < 0.0001) (Cortes et al., Reference Cortes, Saglio, Kantarjian, Baccarani, Mayer, Boqué, Shah, Chuah, Casanova, Bradley-Garelik, Manos and Hochhaus2016). There was also less frequent progression to BP-CML. Rates of grade 3 or 4 haematologic adverse events were higher for dasatinib versus imatinib. The UK STI571 Prospective International Randomised Trial 2 (SPIRIT2) phase 3 clinical trial confirmed these results, with 12-month MMR being achieved in 57.5% versus 46% in the dasatinib and imatinib arms, respectively (p < 0.001) (Osborne et al., Reference Osborne, O’Brien, Hedgley, Foroni, Apperley, Terril, Rowe, McCullough, Holyoake, Pocock, Byrne, Bescoby, Copland and Clark2015). Again, significant treatment toxicities were evident with dasatinib treatment, with pleural effusions occurring more frequently with dasatinib than with imatinib (24.1% versus 1.2%) (Table 3) (Osborne et al., Reference Osborne, O’Brien, Hedgley, Foroni, Apperley, Terril, Rowe, McCullough, Holyoake, Pocock, Byrne, Bescoby, Copland and Clark2015). In patients intolerant or resistant to imatinib, dasatinib has demonstrated durable response rates, with the START-C phase 2 study demonstrating a 2-year CCyR rate of 53%, with 90% of these being maintained at 24 months. Furthermore, the START-R study demonstrated significantly higher rates of CCyR comparing dasatinib 70 mg twice daily with high-dose imatinib at 2 years (i.e., 400 mg twice daily) (44% versus 18%, p = 0.0025) (Kantarjian et al., Reference Kantarjian, Hochhaus, Saglio, de Souza, Flinn, Stenke, Goh, Rosti, Nakamae, Gallagher, Hoenekopp, Blakesley, Larson and Hughes2009; Milojkovic et al., Reference Milojkovic, Apperley, Gerrard, Ibrahim, Szydlo, Bua, Reid, Rezvani, Foroni, Goldman and Marin2012).
Nilotinib, a structural analogue to imatinib, has increased affinity for BCR::ABL1 in vitro compared to imatinib (Weisberg et al., Reference Weisberg, Manley, Breitenstein, Brüggen, Cowan-Jacob, Ray, Huntly, Fabbro, Fendrich, Hall-Meyers, Kung, Mestan, Daley, Callahan, Catley, Cavazza, Azam, Mohammed, Neuberg, Wright, Gilliland and Griffin2005). Like dasatinib, nilotinib has been shown to induce a sustained haematological and cytogenetic response in more patients, when compared with imatinib. The Evaluating Nilotinib Efficacy and Safety in Clinical Trials – Newly Diagnosed patients (ENESTnd) study was a phase 3, randomised clinical trial comparing two doses of nilotinib (300 mg BD and 400 mg BD) to imatinib (400 mg OD). The use of nilotinib was associated with significantly higher MMR at 12 months compared to imatinib, with limited change between the two doses of nilotinib used (44% versus 43% versus 22% for nilotinib 300 mg BD, nilotinib 400 mg BD and imatinib 400 mg OD, respectively; p < 0.001) (Saglio et al., Reference Saglio, Kim, Issaragrisil, le Coutre, Etienne, Lobo, Pasquini, Clark, Hochhaus, Hughes, Gallagher, Hoenekopp, Dong, Haque, Larson and Kantarjian2010; Kantarjian et al., Reference Kantarjian, Hochhaus, Saglio, de Souza, Flinn, Stenke, Goh, Rosti, Nakamae, Gallagher, Hoenekopp, Blakesley, Larson and Hughes2011). Five-year follow-up demonstrated the cumulative incidence of MMR to be 77% for both doses of nilotinib and 60% with imatinib (p < 0.0001) (Saglio et al., Reference Saglio, Kim, Issaragrisil, le Coutre, Etienne, Lobo, Pasquini, Clark, Hochhaus, Hughes, Gallagher, Hoenekopp, Dong, Haque, Larson and Kantarjian2010; Kantarjian et al., Reference Kantarjian, Hochhaus, Saglio, de Souza, Flinn, Stenke, Goh, Rosti, Nakamae, Gallagher, Hoenekopp, Blakesley, Larson and Hughes2011). Although event-free survival was not statistically changed between trial arms, there was an advantage with reducing rates of disease progression in those with intermediate and high-risk disease receiving nilotinib, as estimated through the Sokal score. This highlights the role for second-generation TKIs in prevention of disease transformation. As with dasatinib, adverse events and toxicity with nilotinib use are significant. Within the ENESTnd study, the 6-year cumulative cardiovascular side effect event (CVE) rate was significantly increased in the nilotinib arm, with a dose-dependent effect (Hochhaus et al., Reference Hochhaus, Saglio, Hughes, Larson, Kim, Issaragrisil, le Coutre, Etienne, Dorlhiac-Llacer, Clark, Flinn, Nakamae, Donohue, Deng, Dalal, Menssen and Kantarjian2016). CVEs were reported in 46 (16.5%), 65 (23.5%) and 10 (3.6%) patients, respectively, in the nilotinib 300-mg twice daily, nilotinib 400-mg twice daily and imatinib arms. Sub-analysis demonstrated higher baseline Framingham general cardiovascular risk scores were predictive of patients’ risk of developing a CVE during treatment (Table 3) (Kantarjian et al., Reference Kantarjian, Hughes, Larson, Kim, Issaragrisil, le Coutre, Etienne, Boquimpani, Pasquini, Clark, Dubruille, Flinn, Kyrcz-Krzemien, Medras, Zanichelli, Bendit, Cacciatore, Titorenko, Aimone, Saglio and Hochhaus2021).
Bosutinib is a dual SRC/ABL TKI that demonstrates more potent in vitro activity against BCR::ABL1 than imatinib but has less activity against c-KIT and PDGFR (Puttini et al., Reference Puttini, Coluccia, Boschelli, Cleris, Marchesi, Donella-Deana, Ahmed, Redaelli, Piazza, Magistroni, Andreoni, Scapozza, Formelli and Gambacorti-Passerini2006). The phase 3 clinical trial, bosutinib efficacy and safety in newly diagnosed chronic myeloid leukaemia (BELA), compared the efficacy and safety of bosutinib against imatinib (Cortes et al., Reference Cortes, Kim, Kantarjian, Brümmendorf, Dyagil, Griskevicius, Malhotra, Powell, Gogat, Countouriotis and Gambacorti-Passerini2012). Following a dose escalation to 500 mg OD, the trial allowed the potential for a further increase to 600 mg OD for patients with inadequate molecular and cytogenetic response. At 24 months, cumulative CCyR was similar in bosutinib (79%) and imatinib (80%) arms; however, cumulative MMR rates were significantly higher with bosutinib (59% versus 49%) (Brümmendorf et al., Reference Brümmendorf, Cortes, de Souza, Guilhot, Duvillié, Pavlov, Gogat, Countouriotis and Gambacorti-Passerini2015). Importantly, bosutinib appeared to retain activity across mutations that confer imatinib resistance with the exception of T315I and responses were independent of whether patients had resistance or intolerance to imatinib (Nakaseko et al., Reference Nakaseko, Takahashi, Ishizawa, Kobayashi, Ohashi, Nakagawa, Yamamoto, Miyamura, Taniwaki, Okada, Kawaguchi, Shibata, Fujii, Ono and Ohnishi2015). These results were also supported by the BFORE study which looked at 536 patients with newly diagnosed CP-CML assigned to receive 400 mg of bosutinib once daily (n = 268) or imatinib (n = 268). Achievement of MMR at 12 months was 47.2% versus 36.9%, respectively (p= 0.02) (Cortes et al., Reference Cortes, Gambacorti-Passerini, Deininger, Mauro, Chuah, Kim, Dyagil, Glushko, Milojkovic, le Coutre, Garcia-Gutierrez, Reilly, Jeynes-Ellis, Leip, Bardy-Bouxin, Hochhaus and Brümmendorf2018).
Ponatinib is a multi-targeted kinase inhibitor and is currently the only approved TKI active against the T315I BCR-ABL kinase domain mutation (see below; O’Hare et al., Reference O’Hare, Shakespeare, Zhu, Eide, Rivera, Wang, Adrian, Zhou, Huang, Xu, Metcalf, Tyner, Loriaux, Corbin, Wardwell, Ning, Keats, Wang, Sundaramoorthi, Thomas, Zhou, Snodgrass, Commodore, Sawyer, Dalgarno, Deininger, Druker and Clackson2009), and therefore is an important treatment modality in BCR::ABL1-positive leukaemias. The Ponatinib Ph-positive ALL and CML Evaluation (PACE) trial evaluated 449 patients that were considered resistant or intolerant of second-generation TKIs or harboured T315I mutations (Cortes et al., Reference Cortes, Kim, Pinilla-Ibarz, le Coutre, Paquette, Chuah, Nicolini, Apperley, Khoury, Talpaz, DiPersio, DeAngelo, Abruzzese, Rea, Baccarani, Müller, Gambacorti-Passerini, Wong, Lustgarten, Rivera, Clackson, Turner, Haluska, Guilhot, Deininger, Hochhaus, Hughes, Goldmand, Shah, Kantarjian and PACE2013). The trial used a dose of 45 mg OD ponatinib and patients were stratified by both disease phase and mutational status. At 12 months, major cytogenetic response (MCyR) was achieved in 56% of the patient cohort, with 75% of patients harbouring the T315I mutation achieving MCyR. Long-term follow-up confirmed a sustained response, with 83% of these patients remaining in MCyR at 3 years and 39% of patients achieving MMR or better (Cortes et al., Reference Cortes, Kim, Pinilla-Ibarz, le Coutre, Paquette, Chuah, Nicolini, Apperley, Khoury, Talpaz, DiPersio, DeAngelo, Abruzzese, Rea, Bacca-rani, Muller, Gambacorti-Passerini, Lustgarten, Rivera, Clackson, Turner and Haluska2014). As with other TKIs, significant adverse events were noted. Particularly, arterial occlusive events occurred in 28% of patients (Cortes et al., Reference Cortes, Kim, Pinilla-Ibarz, le Coutre, Paquette, Chuah, Nicolini, Apperley, Khoury, Talpaz, DiPersio, DeAngelo, Abruzzese, Rea, Bacca-rani, Muller, Gambacorti-Passerini, Lustgarten, Rivera, Clackson, Turner and Haluska2014). Through its multi-kinase inhibitory properties, including inhibition of vascular endothelial growth factor (VEGF), it has been postulated ponatinib causes endothelial dysfunction and hypertension, and can promote proatherogenic surface adhesion receptors thus increasing the risk of vascular occlusive events (Table 3; Chan et al., Reference Chan, Talati, Isenalumhe, Shams, Nodzon, Fradley, Sweet and Pinilla-Ibarz2020). The OPTIC trial looked at ponatinib dose ranging to identify optimal benefit/risk outcomes; 45 mg, 30 mg and 15 mg starting doses were used with those in the 45 mg and 30 mg cohort being required to have their dose reduced to 15 mg once daily upon achievement of ≤1% BCR::ABL1. Rates of ≤1% BCR::ABL1 at 12 months were 44.1%, 29.0% and 23.1%, respectively, with subgroup analysis demonstrating poorer response in those with a T315I mutation. However, in those achieving response, this was maintained with dose reduction in 73.3% and 78.6% of patients in the 45 mg and 30 mg cohorts, respectively; with most of those losing response demonstrating the T315I mutation at baseline. The trial concluded that to maximise response while minimising toxicity a 45 mg starting dose reduced to 15 mg on achieving BCR::ABL1 ≤ 1% is recommended (Cortes et al., Reference Cortes, Apperley, Lomaia, Moiraghi, Undurraga Sutton, Pavlovsky, Chuah, Sacha, Lipton, Schiffer, McCloskey, Hochhaus, Rousselot, Rosti, de Lavallade, Turkina, Rojas, Arthur, Maness, Talpaz, Mauro, Hall, Lu, Srivastava and Deininger2021).
TKI resistance – BCR-AB1-dependent and independent mechanisms
BCR-ABL-dependent mechanisms
Point mutations or amplification at the kinase domain of the BCR::ABL1 protein are the most common mechanism by which cells develop resistance to TKIs. More than 90 different BCR::ABL1 kinase domain point mutations have been identified, with a lesser number biologically characterised (Azevedo et al., Reference Azevedo, Reichert, Afonso, Alberca, Tavares and Lima2017).
BCR::ABL kinase domain mutations include those that affect the P-loop, SH2 domain, SH3 domain and the activation loop (Branford et al., Reference Branford, Rudzki, Walsh, Parkinson, Grigg, Szer, Taylor, Herrmann, Seymour, Arthur, Joske, Lynch and Hughes2003; Soverini et al., Reference Soverini, Colarossi, Gnani, Rosti, Castagnetti, Poerio, Iacobucci, Amabile, Abruzzese, Orlandi, Radaelli, Ciccone, Tiribelli, di Lorenzo, Caracciolo, Izzo, Pane, Saglio, Baccarani and Martinelli2006). Mutation type can be associated with phase of disease; M244, M351 and G250 are frequently detected in CP, and mutations at T315, E255 and Y253 are typically found in BP (Soverini et al., Reference Soverini, Colarossi, Gnani, Rosti, Castagnetti, Poerio, Iacobucci, Amabile, Abruzzese, Orlandi, Radaelli, Ciccone, Tiribelli, di Lorenzo, Caracciolo, Izzo, Pane, Saglio, Baccarani and Martinelli2006; Branford et al., Reference Branford, Melo and Hughes2009). The T315I mutation, resulting in the replacement of threonine by isoleucine at the ABL amino acid position 315, is of particular clinical relevance as ponatinib remains the only therapeutic option. It is also important to note that not all mutations result in TKI resistance. Identification of specific mutations is therefore imperative to guide TKI choice (Hehlmann, Reference Hehlmann2020; Smith et al., Reference Smith, Apperley, Milojkovic, Cross, Foroni, Byrne, Goringe, Rao, Khorashad, de Lavallade, Mead, Osborne, Plummer, Jones and Copland2020).
The ELN recommends screening for mutations in any patient with treatment failure or warning response, including a rise in BCR::ABL1 transcript levels, to allow for early detection of mutations and appropriate change in therapy (Hehlmann, Reference Hehlmann2020).
BCR-ABL-independent mechanisms
BCR-ABL1-independent mechanisms of resistance are also described, however, remain poorly understood. These typically result from alterations of cellular signalling and cell cycle regulation downstream of the BCR::ABL1 protein (Li and Li, Reference Li and Li2007). BCR::ABL1 activates alternative signalling pathways, including SRC kinases, RAS and JAK–STAT (Gallipoli et al., Reference Gallipoli, Cook, Rhodes, Hopcroft, Wheadon, Whetton, Jørgensen, Bhatia and Holyoake2014). Aberrant activation of these pathways leads to the CML cell’s proliferation and enhanced survival. The activation of the SRC family kinases has been shown to promote both disease progression, as well as TKI-unresponsiveness. BCR::ABL1 directly interacts with SRC family kinases resulting in both a conformational change in the ABL kinase domain through phosphorylation of SH2 and SH3, as well as activation of SRC family kinases, Hck, Lyn and Fyn, leading to cell growth, differentiation and survival (Stanglmaier et al., Reference Stanglmaier, Warmuth, Kleinlein, Reis and Hallek2003; Hu et al., Reference Hu, Swerdlow, Duffy, Weinmann, Lee and Li2006; Meyn et al., Reference Meyn, Wilson, Abdi, Fahey, Schiavone, Wu, Hochrein, Engen and Smithgall2006). Additionally, while there have been no clinical examples of SRC-activating mutations in imatinib-resistant cell lines or primary CML specimens, cellular activation of this pathway through numerous other crosstalk networks may still facilitate resistance (Donato et al., Reference Donato, Wu, Stapley, Gallick, Lin, Arlinghaus and Talpaz2003; Ptasznik et al., Reference Ptasznik, Nakata, Kalota, Emerson and Gewirtz2004; Wu et al., Reference Wu, Meng, Kong, Peng, Ying, Bornmann, Darnay, Lamothe, Sun, Talpaz and Donato2008). RAS signalling is also activated through Grb2-mediated binding of the Y177 moiety in the BCR sequence, resulting in mitogen-activated protein kinases activation (Cortez et al., Reference Cortez, Reuther and Pendergast1997).
Up-regulation of intracellular efflux transporters and down-regulation of influx transporters has also been demonstrated to confer resistance (Sattler et al., Reference Sattler, Mohi, Pride, Quinnan, Malouf, Podar, Gesbert, Iwasaki, Li, van Etten, Gu, Griffin and Neel2002). Grb2 can recruit Gab-2 with subsequent activation of both PI3k and ERK pathways (Sattler et al., Reference Sattler, Mohi, Pride, Quinnan, Malouf, Podar, Gesbert, Iwasaki, Li, van Etten, Gu, Griffin and Neel2002). The mechanism by which these signalling pathways are activated has been shown to be through the expression of plasma membrane transporter molecules, including the ABC family of transporters, ABCG2 and MDR-1. It has been suggested that over-expression of these protein complexes might confer resistance through reducing intracellular imatinib concentration (Mahon et al., Reference Mahon, Belloc, Lagarde, Chollet, Moreau-Gaudry, Reiffers, Goldman and Melo2003). However, a clear correlation between transporter molecule expression and patient response to imatinib has not been fully elucidated (Mahon et al., Reference Mahon, Belloc, Lagarde, Chollet, Moreau-Gaudry, Reiffers, Goldman and Melo2003).
More recently, greater understanding of the leukaemic stem cell (LSC) and its role in TKI resistance has become apparent. LSCs have been shown to utilise multiple cell-intrinsic pathways, together with microenvironmental and immune cell interactions to evade current therapies including TKIs and have been linked to treatment-refractoriness and disease progression (Hsieh et al., Reference Hsieh, Kirschner and Copland2021). Mitochondrial oxidative phosphorylation within CML LSCs has been well described. Mitochondrial reactive oxygen species are increased in quiescent CML LSCs resulting in increased oxidative DNA damage, genetic instability and clonal evolution (Nieborowska-Skorska et al., Reference Nieborowska-Skorska, Kopinski, Ray, Hoser, Ngaba, Flis, Cramer, Red-dy, Koptyra, Penserga, Glodkowska-Mrowka, Bolton, Holyoake, Eaves, Cerny-Reiterer, Valent, Hochhaus, Hughes, van der Kuip, Sattler, Wiktor-Jedrzejczak, Richardson, Dorrance, Stoklosa, Williams and Skorski2012). Additionally, dysregulation of apoptosis through up-regulation of the BH3 family of proteins including the antiapoptotic proteins MCL-1, BCLxL and BCL2 leads to persistence of LSCs despite treatment (Vetrie et al., Reference Vetrie, Helgason and Copland2020). Carter and colleagues have recently demonstrated eradication of the CD34+ quiescent progenitor cells from BP-CML patient samples using the BCL2 inhibitor, venetoclax, in combination with TKIs (Carter et al., Reference Carter, Mak, Mu, Zhou, Mak, Schober, Leverson, Zhang, Bhatia, Huang, Cortes, Kantarjian, Konopleva and Andreeff2016). Furthermore, clinical trials evaluating BCL2 inhibitors in combination with TKI are currently underway (Jabbour and Kantarjian, Reference Jabbour and Kantarjian2020).
Epigenetic methylation and post-translation acetylation alter gene expression and subsequent protein function. Within CML, the role of epigenetic modification at a biological and therapeutic level are becoming of increasing interest (Scott et al., Reference Scott, Korfi, Saffrey, Hopcroft, Kinstrie, Pellicano, Guenther, Gallipoli, Cruz, Dunn, Jorgensen, Cassels, Hamilton, Cros-san, Sinclair, Holyoake and Vetrie2016). Pre-leukaemic mutations in epigenetic regulators (DNMT3A, TET1, TET2, IDH1 and IDH2) provide the most obvious underlying basis for epigenetic re-patterning in CML (Jan et al., Reference Jan, Snyder, Corces-Zimmerman, Vyas, Weissman, Quake and Majeti2012; Shlush et al., Reference Shlush, Zandi, Mitchell, Chen, Brandwein, Gupta, Kennedy, Schimmer, Schuh, Yee, McLeod, Doedens, Medeiros, Marke, Kim, Lee, McPherson, Hudson, AMK Yousif, Trinh, Stein, Minden, Wang and Dick2014). These changes can lead to functional changes with up-regulation of mitochondrial metabolism and antiapoptotic effects leading to persistence of LSCs despite optimal treatment (Holyoake and Vetrie, Reference Holyoake and Vetrie2017; Vetrie et al., Reference Vetrie, Helgason and Copland2020).
STAMP inhibitors
Asciminib is the most recent drug to be approved for the treatment of CML and is a breakthrough for the small proportion of patients whose disease fails to respond to two or more TKIs. Asciminib is a first in class BCR::ABL1 STAMP inhibitor. Wild-type ABL has a myristoylated N-terminus which is not present in the BCR::ABL1 fusion oncoprotein. Under normal circumstances binding of the terminus to an allosteric site leads to reduced activity, however, when absent as in the BCR::ABL1 fusion protein, this regulation is lost and the protein is constitutively active. Asciminib binds to the allosteric site inhibiting protein activity (Schoepfer et al., Reference Schoepfer, Jahnke, Berellini, Buonamici, Cotesta, Cowan-Jacob, Dodd, Drueckes, Fabbro, Gabriel, Groell, Grotzfeld, Hassan, Henry, Iyer, Jones, Lombardo, Loo, Manley, Pelle, Rummel, Salem, Warmuth, Wylie, Zoller, Marzinzik and Furet2018). Very recently, the phase 3 ASCEMBL clinical trial compared asciminib 40 mg twice daily with bosutinib 500 mg once daily in patients that had failed two or more TKIs, and demonstrated MMR of 25.5% and 12.2%, respectively (p = 0.029). There were also fewer discontinuations with asciminib, 5.8% compared to 21.1% (Réa et al., Reference Réa, Mauro, Boquimpani, Minami, Lomaia, Voloshin, Turkina, Kim, Apperley, Abdo, Fogliatto, Kim, le Coutre, Saussele, Annunziata, Hughes, Chaudhri, Sasaki, Chee, Garcia-Gutierrez, Cortes, Aimone, Allepuz, Quenet, Bedoucha and Hochhaus2021). Asciminib is now approved for third or later line use in patients with TKI resistance or intolerance.
Choosing the right therapy
First-line therapy for CP-CML is invariably TKI therapy, with imatinib, dasatinib, nilotinib or bosutinib all approved for this indication. Generic imatinib is now deemed the most cost-effective first-line therapy in most patients. Although faster responses are seen with the second-generation TKIs, randomised trials with 5- and 10-year follow-up demonstrated no survival benefit (Cortes et al., Reference Cortes, Saglio, Kantarjian, Baccarani, Mayer, Boqué, Shah, Chuah, Casanova, Bradley-Garelik, Manos and Hochhaus2016; Hochhaus et al., Reference Hochhaus, Saglio, Hughes, Larson, Kim, Issaragrisil, le Coutre, Etienne, Dorlhiac-Llacer, Clark, Flinn, Nakamae, Donohue, Deng, Dalal, Menssen and Kantarjian2016). Given the ever-expanding data, it is important to consider patient comorbidities and BCR::ABL1 mutation status when deciding on TKI therapy (Table 4).
Table 4. Decisions on most appropriate therapy, based on BCR::ABL1 kinase domain mutations, relative contra-indications, and current FDA approvals for the different TKIs
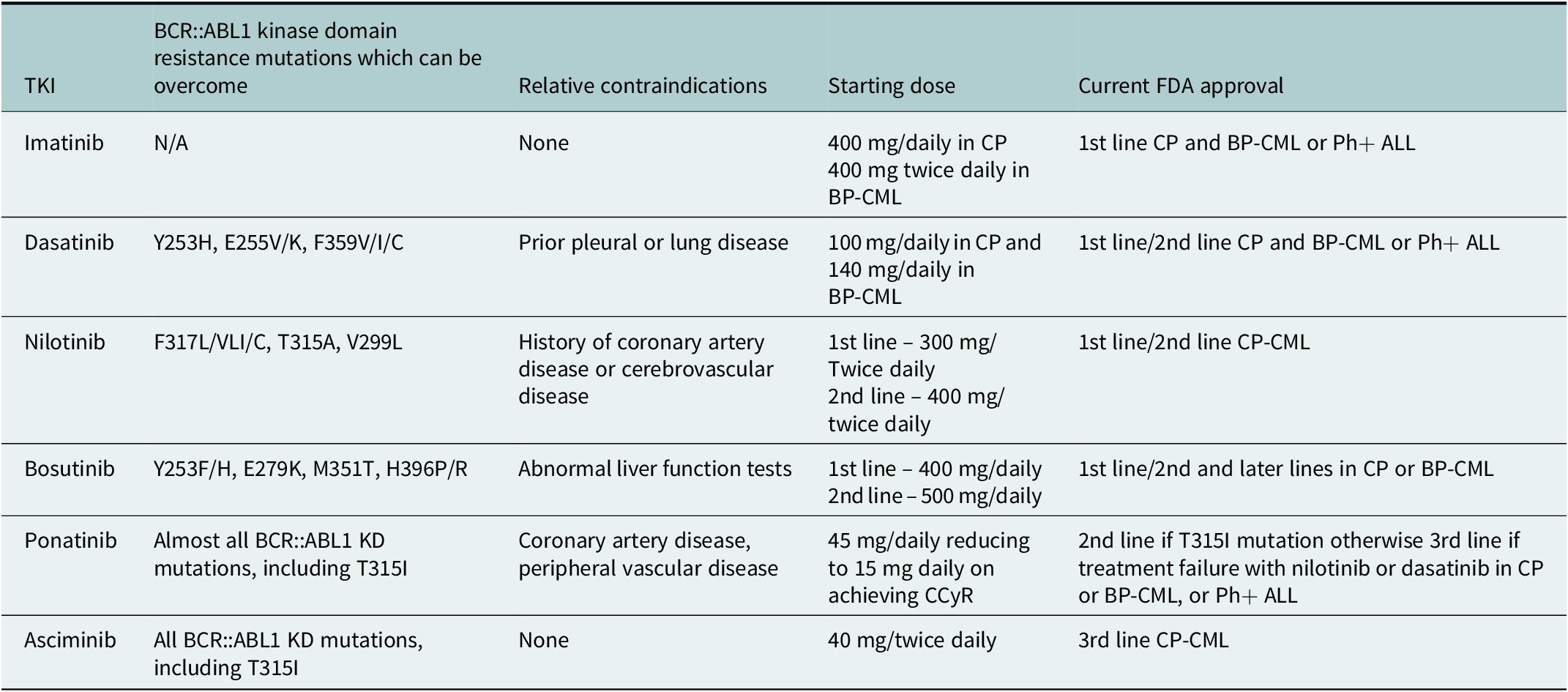
For second-line therapy, where there is intolerance or resistance to first-line TKI, confirmation of compliance is essential in the first instance. Mutational analysis with next-generation sequencing should be performed, with mutations being identified in 4.6% of CP-CML patients and in up to 80% of patients with BP-CML (Branford et al., Reference Branford, Wang, Yeung, Thomson, Purins, Wadham, Shahrin, Marum, Nataren, Parker, Geoghegan, Feng, Shanmuganathan, Mueller, Dietz, Stangl, Donaldson, Altamura, Georgievski, Braley, Brown, Hahn, Walker, Kim, Choi, Park, Kim, White, Yong, Ross, Scott, Schreiber and Hughes2018). Consideration of mutation analysis and patient characteristics, including co-morbidities, should then be made to provide individualised treatment decisions. Ponatinib remains reserved for those patients where a T315I mutation is present or where there has been failure of two previous lines of therapy (Hehlmann, Reference Hehlmann2020; Copland, Reference Copland2022; Copland et al., Reference Copland, Slade, McIlroy, Horne, Byrne, Rothwell, Brock, de La-vallade, Craddock, Clark, Smith, Fletcher, Bishop, Mi-lojkovic and Yap2022).
Despite the significant improvements in outcomes for CP-CML with the advancement in TKI therapy, outcomes for patients presenting in BP or developing BP-CML while receiving TKI therapy remain dismal with alloHSCT being the only treatment option offering the chance of long-term remission or cure. Clonal evolution with acquisition of further oncogenic mutations leads to lymphoid, myeloid or biphenotypic acute leukaemias. Treatment depends primarily on whether BP-CML has developed on treatment. For those fit for alloHSCT without prior TKI exposure, de novo BP treatment is typically with 800 mg imatinib or 140 mg dasatinib, alongside AML or ALL standard chemotherapy approaches, to achieve cytogenetic remission and return to CP before proceeding to HSCT. In those progressing to BP on TKI, the approach is similar, however, ponatinib may be more attractive here. This approach has been supported in the recently published phase 1/2 MATCHPOINT trial which used either 1 or 2 cycles of fludarabine, cytarabine, granulocyte-colony stimulating factor and idarubicin (FLAG-IDA) chemotherapy in combination with ponatinib 30 mg daily before proceeding to alloHSCT with median OS of 12 months and 41% of patients being alive at 3 years (Copland, Reference Copland2022; Copland et al., Reference Copland, Slade, McIlroy, Horne, Byrne, Rothwell, Brock, de La-vallade, Craddock, Clark, Smith, Fletcher, Bishop, Mi-lojkovic and Yap2022).
Dose optimisation and treatment-free remission
Widespread use of TKIs means we now have a plenitude of “real-world” and clinical trial data highlighting the safety of dose modification in maintaining adequate treatment response, while optimising compliance and reducing complications. Prospective clinical trials are still required to aid further improvement to see if we are over-treating optimally responding patients and to support current evidence of dose modifications in patients with intolerable side-effect profiles.
Prospective clinical trials have explored the safety of TKI discontinuation in CP patients with sustained deep molecular remission (DMR; MR4 or better) (Mahon et al., Reference Mahon, Réa, Guilhot, Guilhot, Huguet, Nicolini, Legros, Charbonnier, Guerci, Varet, Etienne, Reiffers and Rousselot2010; Mahon, Reference Mahon2015; Etienne et al., Reference Etienne, Guilhot, Rea, Rigal-Huguet, Nicolini, Charbonnier, Guerci-Bresler, Legros, Varet, Gardembas, Dubruille, Tulliez, Noel, Ianot-to, Villemagne, Carré, Guilhot, Rousselot and Mahon2017). The Stop Imatinib’ (STIM) trial demonstrated an incidence of molecular recurrence at 60 months post cessation of imatinib of 61% (CI 52–70%), with few cases of late recurrence being observed (Mahon et al., Reference Mahon, Richter, Guilhot, Muller, Dietz, Porkka, Hjorth-Hansen, Gruber, Panagoitidis, Ossenkoppele, Mayer, Almeida, Polakova, Ehrencrona, Kairisto, Berger, Olsson stromberg, Mustjoki, Hochhaus and Saussele2014). This suggests that if molecular recurrence is to occur, it happens early. These data are supported by the European Stop Kinase Inhibitor (EURO-SKI) trial (Mahon et al., Reference Mahon, Richter, Guilhot, Muller, Dietz, Porkka, Hjorth-Hansen, Gruber, Panagoitidis, Ossenkoppele, Mayer, Almeida, Polakova, Ehrencrona, Kairisto, Berger, Olsson stromberg, Mustjoki, Hochhaus and Saussele2014, Reference Mahon, Richter, Guilhot, Hjorth-Hansen, Almeida, Janssen, Mayer, Porkka, Panayiotidis, Stromberg, Berger, Diamond, Ehrencrona, Kairisto, Polakova, Mueller, Mustjoki, Hochhaus, Pfirrmann and Saussele2016, Reference Mahon, Richter, Hochhaus, Panayiotidis, de Almeida, Mayer, Hjorth-Hansen, Janssen, Mustjoki, Martinez-Lopez, Vestergaard, Ehrencrona, Kairisto, Dulucq, Machova, Nicolini, Hofmann, Guilhot, Saussele and Pfirrmann2021), which recruited 868 patients of whom 728 were eligible for analysis. Median time on TKI treatment was 7.5 years (range 3.0–14.1 years) with median duration in MR4 before cessation of 4.7 years. 46% of the patients, were still in MMR at 3 years demonstrating cessation of treatment is safe in a significant minority of patients, however, unlike in STIM, late molecular recurrence was more prevalent at 15% between 6 and 36 months emphasising the need for ongoing long-term close monitoring in these patients (Mahon et al., Reference Mahon, Richter, Hochhaus, Panayiotidis, de Almeida, Mayer, Hjorth-Hansen, Janssen, Mustjoki, Martinez-Lopez, Vestergaard, Ehrencrona, Kairisto, Dulucq, Machova, Nicolini, Hofmann, Guilhot, Saussele and Pfirrmann2021). Importantly those patients with molecular recurrence will rapidly re-achieve deep molecular responses on re-starting TKI therapy.
The DESTINY study (Clark et al., Reference Clark, Polydoros, Apperley, Milojkovic, Rothwell, Pocock, Byrne, de Lavallade, Osborne, Robinson, O’Brien, Read, Foroni and Cop-land2019) looked at the role of dose reduction prior to treatment cessation of imatinib, nilotinib or dasatinib in CML and demonstrated that reducing TKI dose to half standard treatment dose was associated with improved tolerability without negative impact on disease control in those with stable MR3 or better. TKI dose was reduced for 12 months prior to cessation of treatment. The trial looked at 49 patients in MMR and 125 in the DMR at the time of dose reduction. In the DMR group, 84 (67%) patients reached the 36-month trial completion point and recurrence-free survival was 72% (95% CI 64–80). In the MMR group, 16 (33%) entrants completed the study and recurrence-free survival was 36% (95% CI 25–53) (Clark et al., Reference Clark, Polydoros, Apperley, Milojkovic, Rothwell, Pocock, Byrne, de Lavallade, Osborne, Robinson, O’Brien, Read, Foroni and Cop-land2019). The higher rates of ongoing DMR in the DESTINY study raise the question of whether initial dose reduction prior to complete cessation of treatment improves outcome. Potential mechanisms are unclear and raise questions around the potential trigger events leading to molecular recurrence. The role of dose reduction on LSC persistence, the bone marrow microenvironment and compliance may all be implicated and requires further work to understand. It does support the argument that treatment cessation for the time being should be reserved for those patients with sustained DMR.
Role in acute leukaemias
The Ph chromosome is the most common cytogenetic abnormality in adults with ALL, being associated with 20–30% of cases. It was historically associated with a poor prognosis, with an increased risk of CNS involvement and an aggressive clinical course. TKIs are now routinely incorporated into standard treatment protocols and have improved outcomes, with initial trials demonstrating a complete response (CR) rate of 92% versus 82% with the addition of imatinib to treatment (Fielding et al., Reference Fielding, Rowe, Buck, Foroni, Gerrard, Litzow, Lazarus, Luger, Marks, McMillan, Moorman, Patel, Paietta, Tallman and Goldstone2014). However, relapse rates remain high and alloHSCT in first cytogenetic remission for those suitable remains key for long-term survival (Liu-Dumlao et al., Reference Liu-Dumlao, Kantarjian, Thomas, O’Brien and Ravandi2012). Paediatric studies have raised the possibility of omitting alloHSCT in the first CR, however, as yet there is insufficient data to incorporate this into adult practice (Ravandi, Reference Ravandi2019). The increasing use of anti-CD19 and anti-CD22 immunotherapy in the form of blinatumomab and inotuzumab ozogamicin, respectively, as well as chimeric antigen receptor T cell therapy (CAR-T) for ALL is likely to change the landscape of managing Ph+ ALL in the coming years. The phase 2 GIMEMA D-ALBA trial looked at the use of blinatumomab in combination with dasatinib with omission of standard cytotoxic chemotherapy as upfront treatment for Ph+ ALL and demonstrated 3-year overall survival of 80% and disease-free survival of 71%. Further work and longer follow up is required, however, this is the first suggestion that it may be possible to omit cytotoxic chemotherapy from Ph+ ALL treatment (Foà et al., Reference Foà, Bassan, Vitale, Elia, Piciocchi, Puzzolo, Canichella, Viero, Ferrara, Lunghi, Fabbiano, Bonifacio, Fracchiolla, di Bartolomeo, Mancino, de Propris, Vignetti, Guarini, Rambaldi, Chiaretti and GIMEMA2020; Curran et al., Reference Curran, Muffly and Luskin2022).
The use of prophylactic TKI therapy to prevent relapse post-alloHSCT has been investigated, however, there remains no firm evidence on which patients will benefit. Guidelines currently recommend close monitoring of marrow and peripheral blood for BCR::ABL1 MRD. All patients should be offered prophylactic treatment for a minimum of 12 months with negative MRD if alloHSCT is performed in CR1 and switched to ponatinib in the event of BCR::ABL1 re-appearance (Ravandi, Reference Ravandi2019).
The choice of TKI remains an area of discussion. Dasatinib has been shown to have increased CNS penetration compared to imatinib, and as such, it has been hypothesised that its use may reduce CNS relapse in particular (Shen et al., Reference Shen, Chen, Cai, Yu, Gao, Hu, Zhai, Liang, Ju, Jiang, Jin, Wu, Wang, Tian, Pan, Jiang, Sun, Fang, Li, Hu, Yang, Zhu, Zhang, Li, Pei, Jeha, Yang, Cheng, Tang, Zhu and Pui2020). Shen et al. published a randomised clinical trial of imatinib versus dasatinib for paediatric patients demonstrating relapse rates of 34.4% and 19.8% (p = 0.01) with CNS relapse rates of 8.4% and 2.7% (p = 0.06), respectively (Shen et al., Reference Shen, Chen, Cai, Yu, Gao, Hu, Zhai, Liang, Ju, Jiang, Jin, Wu, Wang, Tian, Pan, Jiang, Sun, Fang, Li, Hu, Yang, Zhu, Zhang, Li, Pei, Jeha, Yang, Cheng, Tang, Zhu and Pui2020). The European Society for Blood and Marrow Transplantation (EBMT) recommends dasatinib for patients with a prior history of CNS disease. Further research is required to confirm this in a European adult population.
Acute myeloid leukaemia with BCR::ABL1 fusion is a defined genetic entity in the fifth World Health Organization classification (Khoury et al., Reference Khoury, Solary, Abla, Akkari, Alaggio, Apperley, Bejar, Berti, Busque, Chan, Chen, Chen, Chng, Choi, Colmenero, Coupland, Cross, de Jong, Elghetany, Takahashi, Emile, Ferry, Fogelstrand, Fontenay, Germing, Gujral, Haferlach, Harrison, Hodge, Hu, Jansen, Kanagal-Shamanna, Kantarjian, Kratz, Li, Lim, Loeb, Loghavi, Marcogliese, Meshinchi, Michaels, Naresh, Natkunam, Nejati, Ott, Padron, Patel, Patkar, Picarsic, Platzbecker, Roberts, Schuh, Sewell, Siebert, Tembhare, Tyner, Verstovsek, Wang, Wood, Xiao, Yeung and Hochhaus2022). Given the rarity of this condition there is little evidence on the use of TKIs, however, a number of case reports have demonstrated improved outcomes when incorporated into standard treatment. Ph+ AML, even with the advent of TKIs, has a poor prognosis and should be treated with consolidative alloHSCT if CR is reached and this is a viable treatment option (Mizuno et al., Reference Mizuno, Takami, Kawamura, Arai, Kondo, Kawata, Uchida, Marumo, Fukuda, Tanaka, Ozawa, Yoshida, Ota, Takada, Sawa, Onizu-ka, Kanda, Ichinohe, Atsuta and Yanada2021).
Conclusion
Our knowledge of BCR::ABL1-positive leukaemias continues to evolve and with this, a personalised approach to leukaemia management continues to expand. The discovery of TKIs has revolutionised outcomes for patients and provides a key example of 21st century precision medicine. CML management should now be considered as patient-specific, considering patient co-morbidities and lifestyle. For patients with intermediate or high-risk disease, as dictated through the prognostic scoring systems, there should be strong consideration for a second-generation TKIs upfront (Ciftciler and Haznedaroglu, Reference Ciftciler and Haznedaroglu2021).
There remain many unanswered questions and ongoing clinical trials exploring BCL2 inhibitors as well as other agents to specifically target quiescent stem cell populations, and increasing use of STAMP inhibitors for refractory cases are likely to improve outcomes in CML (Jabbour and Kantarjian, Reference Jabbour and Kantarjian2020). Additionally, immunotherapy and CAR-T in Ph+ ALL are expanding areas. Critical areas of unmet need remain, including management of patients with TKI resistance and BP-CML, increasing the number of patients able to attempt and maintain successful treatment-free remission, and improvements in dose optimization and TKI selection to minimise side effects of long-term therapy with further research required.
Open peer review
To view the open peer review materials for this article, please visit http://doi.org/10.1017/pcm.2023.9.
Author contribution
S.L. wrote the first draft of the manuscript. G.A.H. and M.C. edited the manuscript. All authors approved the final version.
Financial support
M.C. and G.A.H. are employees of the University of Glasgow. No additional funding was required for preparation of this review article.
Competing interest
M.C. has received research funding from Cyclacel and Incyte, is/has been an advisory board member for Novartis, Incyte, Jazz Pharmaceuticals, Pfizer and Servier, and has received honoraria from Astellas, Novartis, Incyte, Pfizer and Jazz Pharmaceuticals. G.A.H. and S.L. have no conflicts of interest.





 Open access
Open access
Comments
Dear Laeti and Jess,
Please find attached our submission on Precision Medicine in Chronic Myeloid Leukaemia. I think this is an ideal topic for your journal as CML is one of the first cancers to be very successfully treated with a precision medicine approach. All authors have reviewed the manuscript and agree to submission. All conflicts of interest have been declared.
Many thanks for you assistance with the publication process.
Kind regards
Mhairi Copland