


Introduction
Glioblastoma (GBM) is the most common primary brain tumour that accounts for almost half of all primary brain tumours with an survival rate of 4% at 5 years (Refs Reference Grochans1, Reference Poon2). The worldwide incidence of GBM varies across studies between 0.59 and 5 per 100.000 (Refs Reference Grochans1, Reference Grech3). The incidence is higher in men with a median age at diagnosis of 64 years old (Ref. Reference Tamimi, Juweid and De Vleeschouwer4). GBM is classified as a grade IV aggressive primary brain glioma, and due to advancements in understanding its genomic biology the current WHO classification includes molecular features in defining a GBM, such as the IDH and TERT mutation and EGFR amplification (Ref. Reference Louis5). Several markers such as TP53 mutation, MGMT, TERT and ATRX play an important role in understanding the pathophysiology of GBM, being as well potential targets for future targeted therapies (Refs Reference Grochans1, Reference Tamimi, Juweid and De Vleeschouwer4).
While the next-generation sequencing technologies became more accessible in both research and clinical setups, a more in-depth characterisation of the tumour genomic profile became available (Ref. Reference Mardis6). This allowed the identification of specific genetic anomalies, present in both the coding and non-coding regions of the genome. Therefore, the repository of therapeutic targets expended, supporting the development of new targeted therapies against GBM that are looking at specific genetic event inhibition or are targeting complex molecular pathways such as DDR (Refs Reference Wang7–Reference Rominiyi and Collis9).
The current therapeutic interventions for GBM include maximal surgical resection, radiotherapy (RT) and concomitant or maintenance temozolomide (TMZ) chemotherapy. However, none of them are curative and most of patients will experience multiple recurrences. In fact, regardless of the numerous ongoing clinical trials, the development of effective therapies is deficient due to high intratumor heterogeneity, lack of adequate control arms, selection bias and small sample size (Refs Reference Fernandes and De Vleeschouwer10, Reference Weller11).
Recently, the DNA damage response (DDR) pathway became subject of interest in GBM research after its implications in chemotherapy and RT resistance were better understood given the advanced in understanding of the underling mechanisms (Ref. Reference Ferri12). DDR is involved in detecting the DNA damage, signalling its presence and favouring its repair (Refs Reference Ferri12, Reference Jackson and Bartek13).
Since both, RT and chemotherapy induce DNA damage and activate DDR, molecules that target DDR inhibition are investigated for their therapeutic role in overcoming treatment resistance (Ref. Reference Everix14). Surprisingly, even though the DDR pathway activates post-irradiation in both parent cells and recurrent cells, Kaur et al. demonstrated on GBM cell line models (U87MG and SF268) that there are differences in the DDR pathway activation in primary versus recurrent post-treatment GBM cells. Post-irradiation, recurrent cells prefer a different DDR pathway in comparison to their parents, highlighting the adaptability of GBM cells. As a result, more studies on which DDR enzyme is acting in recurrent GBM should be conducted since the DDR enzymes ATR and ATM have different mechanisms of action (Ref. Reference Kaur15).
DDR are induced as an adaptive reaction to either single or double-strand breaks (SSB/DSB) of the genomic DNA, which occur as a consequence of the endogenous genomic instability associated with GBM development or as a post-conventional treatment side-effect. The restoration of SSB, which are the most common DNA lesions, is mediated by base excision repair (BER), nucleotide excision repair (NER) and mismatch repair (MMR) mechanisms, while the DSB restoration is mediated by either homologous recombination (HR) or nonhomologous end-joining (NHEJ) mechanisms (Refs Reference Bonm and Kesari16, Reference Caldecott17) One clear example for the use of DDR pathway in GBM therapy are the poly ADP-ribose polymerase (PARP) inhibitors, especially those that target PARP-1. Normally, this enzyme binds to the injured DNA site catalysing the formation of ADP-ribose polymers using NAD+ as a substrate and activates the necessary enzymes for BER to be conducted. PARP inhibitors (PARPi) bind themselves to this enzyme, thus blocking the formation of ADP-ribose polymers from NAD+. Therefore, DNA repair is ceased which leads to an increase in genomic instability, growth arrest and apoptosis (Refs Reference Malyuchenko18, Reference Leonetti19). These molecules have started to being investigated in GBM, but with limited results which might be due to increased intratumor heterogeneity that allows for rapid development of treatment resistance subclones or to lack of proper cancer biomarkers 25709118 and 34584069. A possible new source of biomarkers that will allow real-time evaluation of the treatment efficacity and development of resistance is represented by non-coding (ncRNAs) (Refs Reference Sakthikumar20–Reference Zhang and De Vleeschouwer22). Multiple research directions have emerged intending to investigate the expression levels and the regulatory impact of these ncRNAs, especially microRNAs (miRNAs), long non-coding RNAs (lncRNAs) and circular RNAs (circRNA) on the DDR pathways in the context of GBM pathogenesis, chemoresistance and radiation sensitivity. (Refs Reference Zeng23–Reference Parashar, Ravindran and Choudhary27). NcRNAs have been intensively studied in multiple cancers due to their wide tissue distribution, ease of access and potential to be used as biomarkers and treatment targets (Refs Reference Zhang and De Vleeschouwer22, Reference Mousavi28). In GBM dysregulated ncRNAs can be used both to target altered tumour mechanisms using substation strategies or to down-regulate specific miRNAs (Ref. Reference Huang29). Hence, due to their versatile activity as GBM promoting or suppressing agents, ncRNAs started to be investigated as valuable therapeutic targets or as biomarkers to evaluate treatment response.
The subject of DDR inhibitors in the management of GBM patients is a highly studied field with an international effort being in place to advance the available treatment options of GBM using DDR inhibitors. The National Brain Tumour Society is the institution driving these efforts, through establishment of a DDR consortium. The DDR consortium has the mission to move forward to the clinic DDR inhibitors through supporting research, encourage exchange of data and evaluation of clinical trials (Ref. 30).
The aim of this review is to decipher the intricated machinery involved in the regulation of DDR pathway in GBM. Our approach will focus on the functional mechanisms activated by both endogenous and exogenous DNA damage-inducible agents. We will use an innovative approach towards DDR by looking at the regulatory roles of ncRNA molecules and how we can use these molecules both as biomarkers and potential therapeutic targets.
DDR pathway statut in GBM
DNA damage can be induced endogenously, by mutations inducing genomic instability or exogenously by exposure to various harmful factors such as ultraviolet, RT, chemotherapy, reactive oxygen species (ROS) and deregulated metabolism respectively (Refs Reference Weber and Ryan31, 32). Alterations in DNA as SSB or DSB are a common event that led to the development of a complex regulatory network to protect and restore DNA integrity represented by the DDR pathway (Ref. Reference Ciccia and Elledge33). DDR pathway prevents damaged DNA to be copied (G1 checkpoint) or transferred to the future generation of cells (G2 checkpoint) during cellular cycle (Ref. Reference Smith34). The Phosphatidylinositol-3 kinase-like protein (PIKK), includes ataxia–telangiectasia mutated (ATM) and ataxia-telangiectasia Rad3 related (ATR), DNA-Dependent Protein Kinase Catalytic Subunit (DNA-PKs), mammalian target of rapamycin, suppressor of morphogenesis in genitalia and transcription-associated protein (Refs Reference Weber and Ryan31, Reference Woods and Turchi35). ATM, ATR and DNA-PK are the most known kinases involved in DDR, their integrity being instrumental to the pathway regulation and cell survival (Refs Reference Weber and Ryan31, Reference Smith34).
GBM is a highly treatment-resistant brain malignancy and during its treatment with TMZ and RT is supposed to high DNA damage stress including SSB and DSB (Refs Reference Kaina and Christmann36–Reference Aldea38). ATM is activated in response to DSB and plays a role in repairing the DNA damage by recruiting proteins, signalling the cell checkpoint and inducing apoptosis (Ref. Reference Estiar and Mehdipour39). The role of ATM in phosphorylating various compounds (p53, CHK2, H2AX), thus inducing the cell cycle arrest and apoptosis is well-known (Refs Reference Weber and Ryan31, Reference Smith34). The phosphorylation of CHK2, which is the most important ATM transducer, induces the G1 checkpoint arrest and apoptosis (Refs Reference Smith34, Reference Zannini40, Reference van Jaarsveld41). Activated CHK2 activation favours the phosphorylation of CDC25A (one of the most crucial cell cycle regulators) (Ref. Reference Shen and Huang42) which leads to its reduction and blockade of the entrance to the G1 phase (Refs Reference Estiar and Mehdipour39, Reference van Jaarsveld41, Reference Falck43). However, G1 checkpoint arrest is mainly regulated through the p53 pathway (Ref. Reference Jeggo44). Activated p53 induces the transcription of CDKN1A which encodes p21 (Ref. Reference Fridman and Lowe45); p21 mediates the p53-dependent G1 cycle arrest through inhibition of the cyclin-dependent kinases (CDK1 and CDK2) (Refs Reference Smith34, Reference Abbas and Dutta46). TP53 mutations are very frequent in GBM (up to 70%) and are associated with increased tumour progression and the inactivation of p53 is associated with an aggressive phenotype and sustained cell viability (Ref. Reference Lee47).
ATR is activated by various types of DNA damage, including DSB, cross-links and DNA replication stress (Ref. Reference Nam and Cortez48). Compared to ATM, ATR is an essential component of the cell for its viability and replication (Ref. Reference Sirbu and Cortez49). Studies showed that at the N-terminal, ATR presents a binding protein (ATRIP); the complex ATR/ATRIP is mediated through RPA (Replication protein A) to initiate the DDR by phosphorylating various targets, including CHK1 (Ref. Reference Rao50). Further, CHK1 phosphorylates CDC25 proteins (A, B, C) to arrest the G2 checkpoint (Refs Reference Smith34, Reference Zannini40, Reference Saldivar51). Moreover, it was shown that ATR/CHK1 plays a role in the intra-S-phase cell cycle checkpoint by phosphorylating a Treslin protein that blocks the accumulation of CDC45, an important protein for initiation of DNA replication (Refs Reference Weber and Ryan31, Reference Zannini40, Reference Saldivar51, Reference Guo52). Wee1 is a kinase activated by CHK1 and is ‘the gatekeeper’ of the G2 checkpoint and S phase, arresting mitosis (Fig. 1). It favours the phosphorylation of CDK1, thus its inactivation during interphase. Studies have shown that the downregulation of Wee1 was associated with an increased entry in the mitotic phase (Ref. Reference Esposito53).
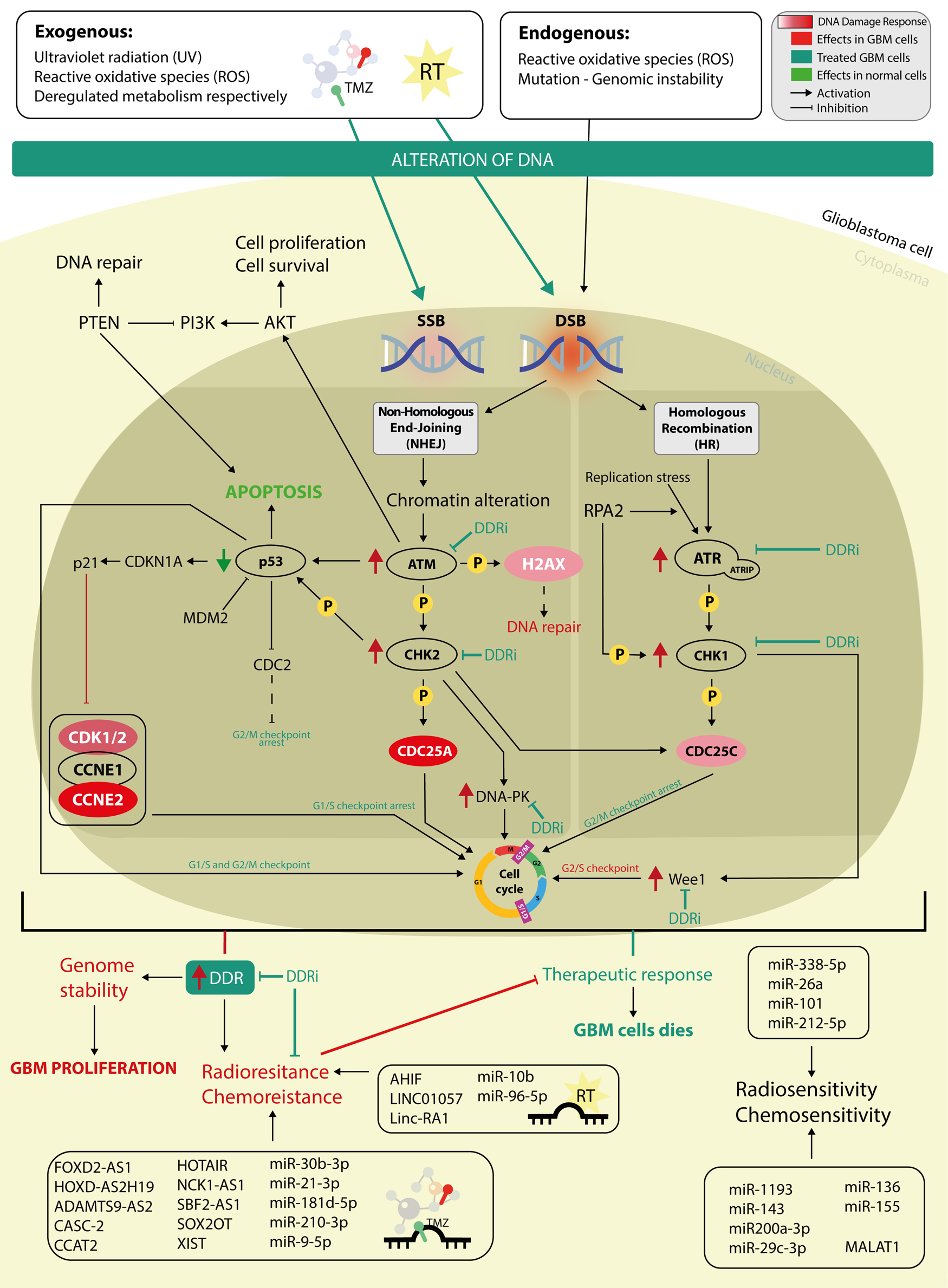
Fig. 1. The intricate mechanism of DDR pathway in glioblastoma with a focus on miRNAs and lncRNAs involved in the regulation of radioresistance and chemoresistance. DNA is damaged by exogenous and enogenous factors and is repaired by two principal repatory pathways, non-homologous end-joining (NHEJ) and homologous recombination, both which can be altered in the development of glioblastoma at different key points. Targeting an altered DDR pathway using DDR inhibitors (DDRi) represents an attractive treatment approach. The main molecules targeted by DDRi are represented by: ATM, ATR, Wee1, CHK1 and CHK2.
The above-mentioned DDR pathways represent specific targets that could be used for future therapy development (Fig. 1) (Ref. Reference Rominiyi and Collis9).
DNA damage response induced by Temozolomide and Radiotherapy
Activation of the DDR pathway is a common mechanism for cancer induced chemotherapy or RT resistance which limit current therapeutic approaches (Ref. Reference Weber and Ryan31). TMZ is an alkylated agent, part of the triazene group compounds (Ref. Reference Strobel54). In 2005, it became the standard of care, together with surgery and RT in GBM, by showing prolongation of survival by 2.5 months when compared to RT alone. TMZ by alkylating the DNA, forms two compounds: N3-methyladenine and N7-methylguanine (~90%) and O6-methylguanine (5–10%) (Ref. Reference Lee55). O6-methylguanine(O6MeG) represents an important signal for generating DDR (Ref. Reference Cui56). O6-methylguanine-DNA methyltransferase (MGMT) is an endogenous enzyme that contributes to DDR by removing the methyl group in O6-methylguanine thereby neutralising the drug-induced DNA damage and reducing the overall efficacy of TMZ (Ref. Reference Singh57). MGMT is a well-known factor that contributes to TMZ resistance in GBM, yet not the only factor responsible for TMZ resistance (Refs Reference Kitange58, Reference Chien59). For example, it was shown that GBM with a low level of MGMT had increased rho-associated kinase 2 (ROCK2), a cytoskeleton regulator, that was associated with a low survival rate, making it a potential target for future treatments (Ref. Reference Zhang60). O6MeG plays a role also in the MMR pathway damaging the DNA replication (Ref. Reference Rominiyi and Collis9).
Besides the afore-mention mechanism, MMR might influence the resistance to TMZ and also thorough development of de novo mutations or though MMR deficiency (Refs Reference Singh57, Reference Touat61). This pathways includes mainly 4 proteins (MLH-1, MSH-2, MSH-6 and PMS-2) which act as endonucleases (MSH-2 and MSH 6) or are signalling the initiation of repairment (MSH −1/PMS-2) (Ref. Reference Bateman62). The expression of these proteins can be routinely detected through immunohistochemistry (IHC). Their lack of expression is associated with a MMR deficient mechanism, microsatellite instable (Ref. Reference McCarthy63).
RT is used as a standard treatment for GBM in shrinking the mass or post-surgery to eliminate the residuals (Refs Reference Borrego-Soto64, Reference Gzell65). RT induces a various types of DNA damage, directly by inducing DSB and indirectly by promoting accumulation of ROS which favours SSB (Ref. Reference Borrego-Soto64). In the SSB, PARP1 plays a central role in the detection and repair of the pathway (Ref. Reference Rominiyi and Collis9). SSB and base modification form multiple DNA lesions, favouring a change in base-pairing properties and causing spontaneous mutations (Ref. Reference Caldecott17). PARP 1 bounds SSB and PAR. The X-ray repair cross-complementing protein 1 (XRCC1), acts as a scaffold for SSB break proteins, thereby stimulating the repair process (Ref. Reference Ray Chaudhuri and Nussenzweig66). By targeting PARP and ATR pathway, the response to RT increase (Ref. Reference Carruthers67).
DSBs are repaired through two mechanisms, the NHEJ and HR DNA repair pathways (Ref. Reference Rominiyi and Collis9). The HR mechanisms takes place in late G1 and G2 phases and is using the sister chromatid as a DNA replication template to correct the damage (Refs Reference Rominiyi and Collis9, Reference Krajewska68).
RAD51 is a specific protein that has a role in DNA damage repair (DDR) through HR (Refs Reference Hine69, Reference Balacescu70). It is overexpressed in multiple cancers, including glioblastoma, and is associated with resistance to treatment. Furthermore, in various studies, its overexpression was associated with genetic mutations that favoured the evolution of the tumour and even metastasis (Ref. Reference Gachechiladze71). In a study by Morrison et al., they showed that RAD51 in the HR pathway was overexpressed in patients with GBM versus normal brain, and it was associated with a reduced survival rate, making RAD51 a potential prognostic biomarker (Ref. Reference Morrison72). In NHEJ, Ku70–Ku80 hetero dimer (Ku) plays a central role in detecting the DSB and behaves as a loading protein on which other NHEJ proteins can be recruited as needed to promote the joining of DNA ends. The NHEJ pathway utilises proteins that have various roles from recognising to ligating the DNA ends in a flexible manner (Ref. Reference Chang73). Both, HR and NHEJ are regulated by CDK activity (Ref. Reference Rominiyi and Collis9).
The Fanconi anaemia pathway is activated by both ATR and ATM kinases and has a role in facilitating lesion repair and restarting the replication via NER, HR- and MMR system (Fig. 2) (Ref. Reference Rominiyi and Collis9).
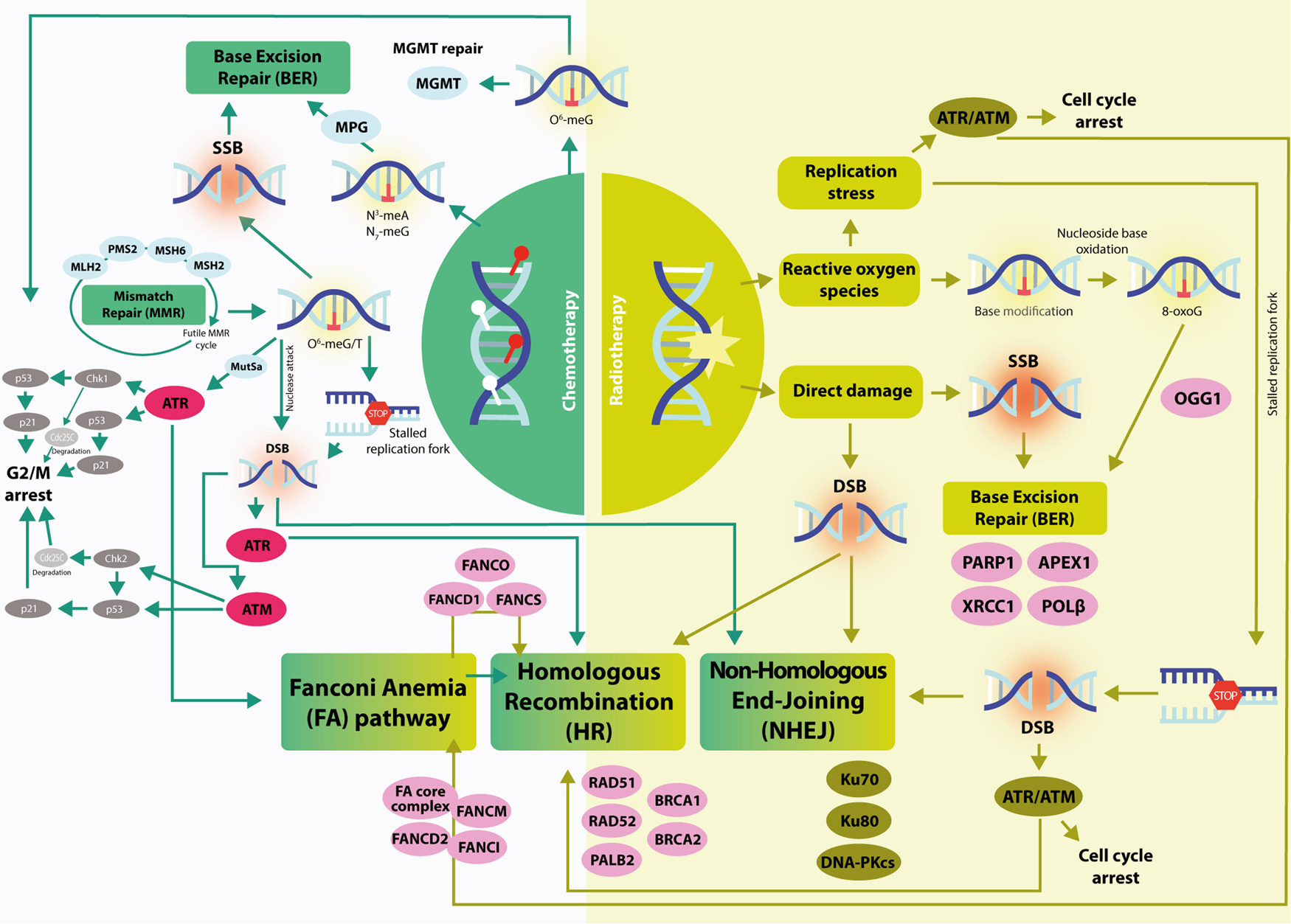
Fig. 2. A graphical representation of the main DDR mechanisms activated by RT (right) and TMZ (left) in GBM. Right: RT induce direct damage, causing SSB and DSB; they are repaired by BER and DSB repair pathways (HR and NHEJ). Replication stress activates the ATR/ATM kinases, thus the cell cycle arrest (see Fig. 1). Stalled replication forks results from replication stress and are repaired via activation of the FA pathway. The detection of SSB and its repair is made by PARP1, together with XRCC1. ROS induced by RT cause base modification which is repair by BER. Left: TMZ induces O6-MeG, which is repaired by MGMT or it can undergo the MMR cycle if an aberrant mutation is present (O6MeG/T). The O6-meG/T lesion can either be recognised by ATR and lead to cell cycle arrest and DNA repair, resulting in a stalled replication fork or form a DSB via nuclease attack, thereby activating G2/M arrest through ATM. It can be repaired through HR and NHEJ. N3- and N7-meA lesions are recognised and repaired through BER pathway via PARP1.
Biomarkers of DDR pathway status
Assessing the level of activity of the DDR pathway is crucial for the identification of specific biomarkers that can predict the response to specific classes of DDR inhibitors (Ref. Reference Chen74). Among the first proposed biomarkers is the status of the TP53 gene which is altered in up to 20–30% of primary GBM and more than 60% of secondary GBM (Ref. Reference England75). Intact p53 activity is required for efficient cell cycle progression through the G1/S checkpoint, its mutational loss of function is allowing cancer cells to pass the checkpoints with replicating errors and to accumulate DNA damage (Ref. Reference Bonm and Kesari16). For example, treatment with RT and DNA-PK inhibitor M3814 on p53 mutated cell lines lead to mitotic catastrophe and apoptotic cell death, as the replication mechanism was unable to arrest the cell cycle and repair the induced DNA damage (Ref. Reference Sun76). Also, dysfunction of p53 is influencing the cell response to the G2/M checkpoint, therefore increasing cellular susceptibility to ATR, CHK2 and Wee1 inhibitors (Ref. Reference Bonm and Kesari16).
Mutated IDH1/2 enzymes will impact the citric acid cycle leading to the generation of increased quantities of 2-hydroxyglutarate, an oncometabolite that induces aberrant hypermethylation of histone 3 lysine 9 (H3K9) which impacts the efficacy of DDR proteins, such as p53 binding protein 1 (53BP1) (Ref. Reference Young77). Therefore, mutations in IDH1/2 genes are associated with a dysregulation of the DDR pathway to DSB which sensitises tumours harbouring these mutations to ATR and PARPi (Ref. Reference Sule78).
PTEN is a tumour suppressor gene that is mutated or deleted in various cancers, including in 40% of GBM cases. PTEN is a negative regulator of the PI3 K/AKT signalling pathway, therefore PTEN dysregulation is associated with PI3 K/AKT activation which leads to cancer cell proliferation, failure to arrest the cell cycle at, G2/M, and resistance to classic GBM therapy (Refs Reference Eich79–Reference Benitez81). Loss of PTEN leads to sensitivity to genotoxic stress, accumulation of DNA damage and hyperactivation of ATM. In a cell line model in which PTEN was inhibited using siRNA, treatment with ATM inhibitor KU-60019 caused increased catastrophic DNA damage, mitotic cell cycle arrest and apoptosis when compared with PTEN wild-type cells (Ref. Reference McCabe82). Therefore, PTEN gene loss of function mutation can be investigated as a possible biomarker for response to ATM inhibitors in GBM. Additional strategies focus on targeting RAD51 protein in PTEN deficient GBM models (Ref. Reference Turchick83).
PTEN deficient tumour cells have increased replication stress and rely on the activation of PTEN-RAD51 signalling axis to ensure efficient DNA replication (Ref. Reference He84). RAD51 is a key protein that is activated in conditions of increased DNA damage that acts on correcting errors in the replication forks through HR (Ref. Reference Hine69). Intact function of the RAD51 protein and its paralogs is essential for efficiently bypassing blockades in the replication forks by allowing switching of the replication fork on the sister DNA chromatide (Ref. Reference Rein85). RAD51 IHC can be used as a surrogate marker for the activation of the DDR pathway (Table 1) (Ref. Reference Welsh86). Blocking RAD51 in PTEN deficient cells using an efficient cell-penetrating autoantibody that inhibits RAD51, 3E10, is inducing mitotic catastrophe and subsequent apoptosis (Ref. Reference Turchick83). Furthermore, in various studies, its overexpression was associated with genetic mutations that favoured the evolution of the tumour and even metastasis (Ref. Reference Gachechiladze71).
Table 1. Potential protein biomarkers for the evaluation of the DDR pathway status

Recent advances in the understanding of genome biology and mechanism of the DDR pathways have led to the identification of possible surrogate biomarkers of the DDR pathway activation that can be more easily detected in clinical settings using classic detection methods widely available in pathology laboratories (Table 1) (Refs Reference Pirlog87, Reference Susman88). An example is the phospho-H2AX or γ-H2AX that functions as a sensitive marker for the presence of DNA DSB (Ref. Reference Palla89). Accumulation of γ-H2AX, can be easily detected by both WB and IHC, is a marker of accumulating DNA damage and has been widely investigated for its role as a surrogate for response to agents targeting the DDR pathway (Ref. Reference Nagelkerke and Span90).
DDR and non-coding RNAs in GBM targeted therapy
Despite all the efforts made in optimising the standard regiment of care for GBM, patient outcome has not significantly changed. Emerging experimental findings started to highlight the potential therapeutic benefit of targeting the DDR pathway and its related non-coding components (Refs Reference Rominiyi and Collis9, Reference Ferri12). As such, a growing body of evidence emerged supporting the role of ncRNAs expression in the modulation of DDR genes during the pathogenesis and relapse of GBM, but also their potential utility as reliable biomarkers for targeted therapy (Refs Reference Stackhouse26, Reference DeOcesano-Pereira98–Reference Wu100). Today, ncRNAs are recognised as a diverse group of transcripts that are not translated into proteins following transcription, but rather play a significant role in modulating the expression of several genes involved in different pathological processes, including GBM carcinogenesis, tumour development, metastasis and DDR (Refs Reference Beylerli101–Reference Paulmurugan103). In particular, miRNAs, lncRNAs and circRNAs that have a dysregulated expression profile started to be investigated for their role of DDR mediators in GBM, Moreover, the strong regulatory influence of miRNAs, lncRNAs and circRNA upon the pathogenic properties of glioma cells, including treatment resistance and sensitivity, are making them attractive targets for therapy (Refs Reference Lu104, Reference Visser and Thomas105).
Several research studies confirmed the active involvement of dysregulate ncRNAs expression profiles in the pathogenesis and development of GBM (Refs Reference Chen106–Reference Yadav108). Hence, their utility as therapeutic interventions against GBM is based on the concept that the malignant phenotype can be restored by targeting different ncRNAs. As such, two primary ncRNA-based approaches are currently investigated for the development of anti-GBM therapies: Gene-silencing therapy which uses specific single-stranded oligonucleotides with complementary sequences to inhibit the function of a targeted ncRNA, and replacement therapy which aims to restore the expression of silenced ncRNAs with ncRNA mimics. Therefore, both strategies could aid the development of better ncRNA-based GBM targeted therapeutics (Refs Reference Winkle109–Reference Shetty112).
The repository of dysregulated ncRNAs that are currently explored as potential therapeutic targets against GBM is growing. However, extensive research and validation is required to safely translate such experimental knowledge into efficient clinical applications.
DDR and miRNAs in GBM
In an experimental study conducted by Costa et al., it was demonstrated that intravenously administered chlorotoxin-targeted stable nucleic acid lipid particle-formulated anti-miR-21 oligonucleotides, efficiently promoted miR-21 silencing. Moreover, increased mRNA and protein levels of RhoB, a direct target of miR-21, with no signs of systemic immunogenicity was clearly observed, while decreased GBM cell proliferation and tumour size, enhanced apoptosis and improvement of animal survival (Ref. Reference Costa113). In a similar work by Lee et al., anti-miR-21 was delivered using a multi-valent folate-conjugated three-way-junction-based RNA nanoparticle platform. As a result, anti-miR-21 specifically targeted and knocked down miR-21 expression in GBM cells in vitro and in vivo while also upregulating the expression of PTEN and PDCD3 genes which increased GBM apoptosis and induced tumour regression (Ref. Reference Lee114).
In a distinctive research study, Huang et al. investigated the regulatory effects of miR-93 on the autophagic activity of GSCs revealing that IR and TMZ, two first-line treatments for GBM, decreased miR-93 expression which resulted in enhanced autophagic processes. However, the researchers showed that ectopic miR-93 expression inhibited autophagy and enhanced the activity of IR and TMZ against GSCs (Ref. Reference Huang29).
Replacement therapy has also been validated in the experimental setup. As such, Li et al. reported that overexpression of miR-519a, targeted with miR-519a mimic, enhanced TMZ chemosensitivity and promoted autophagy in GBM by regulating STAT3/Bcl2 signalling pathway (Refs Reference Susman88, Reference Li115). In 2021, Nan et al. enhanced the expression of miRNA-451, a tumour suppressor that is usually suppressed in high grades GBM, using a transfected lentivirus expressing miR-451. This supports the utility of miR-451-targeted therapy for GBM, as its overexpression regulates NF-κB signalling pathway by targeting IKKβ, thus inhibiting tumour cells growth in vitro and in vivo (Ref. Reference Nan116).
The role of miR-490 upon the activation of p53, a well-known master regulator of DDR, was observed by Vinchure et al. while conducting an in vitro telomerase fragility study on GMB cell lines U87MG (wild-type p53) and T98 G (p53 mutant c.711G > T). They have reported that miR-490 overexpression-induced DNA damage and DDR signalling in U87MG cells, with the upregulation of p53 due to an accumulation of p-γH2AX (Refs Reference Mah117–Reference Williams and Schumacher119). Thus, by upregulation of p53, miR-490 overexpression could indirectly orchestrate a variety of DDR mechanisms in GBM cells.
In another study, designed to evaluate the miR-338-5p effect upon radiation response in GBM cells, Besse et al. found that overexpression of miR-338-5p, gained by transient transfection of IR-treated GBM cell lines (A172, T98G, U87MG), lead to downregulation of NDFIP1, RHEB and PPP2R5a. These genes have been previously described as key components of the DDR pathway; thus, this study provides evidence supporting the regulatory effect of miR-338-5p on the IR response phenotype of GBM cells through direct upregulation of DDR genes (Ref. Reference Besse120).
Previous studies reported the miR-181b pathway being activated as a reaction to several DNA lesions, including GBM response to TMZ-induced methylation and IR-induced DSB (Refs Reference Wang121, Reference Soubannier and Stifani122). In a distinct study, Xu et al. reported that overexpression of miR-181b increases IR-induced NF-κB activity by downregulating SENP2 in IR-treated GBM cell lines (T98G, U87MG). Taken together, these observations support the role of miR-181b as a positive regulator on the feedback loop of NF-κB activation via targeting SENP2 in GBM cells exposed to DNA damaging agents, such as chemo and radiotherapies (Ref. Reference Xu123).
Finally, two independent studies focused on exploring the implication of miR-221/miR-222 in the molecular process associated with GBM pathogenesis found different links between these genomic modulators and DDR-mediated treatment response. Li et al. found that radiation-induced c-jun transcription of miR-221/miR-222 modulated DNA-PK expression to affect DDR by activating Akt independent of PTEN status, contributing to a radio-resistance phenotype. Thus, miR-221/222 could serve as a therapeutic target for increasing radiosensitivity in GBM cells (Ref. Reference Li124). Separately, Quintavalle et al. found that miR-221/miR-222 are overexpressed in GBM cells and directly downregulate MGMT in GBM TMZ-resistant cell lines, inducing greater TMZ-mediated cell death. However, as MGMT is a key component of the GBM-associated DDR pathways, miR-221/222-mediated MGMT downregulation may render cells unable to overhaul genetic damage (Ref. Reference Quintavalle125). Taken together, these studies highlight two distinct mechanisms behind the regulation of DRR in GBM and GBM-resistant cell lines.
So far, over 250 miRNAs are known to be upregulated in GBM-associated pathways, including DDR, contributing to the development of either treatment-resistant or treatment-sensitive phenotypes (Refs Reference Banelli99, Reference O'Brien126–Reference Peng and Croce128). Table 2 presents a summary of the principal miRNAs that influence the DDR pathway upon GBM treatment.
Table 2. Overexpressed miRNAs in GBM-associated DDR and the effect upon treatment

DDR and lncRNAs in GBM
Generally, lncRNAs can function as molecular decoys, scaffolds, enhancers or repressors. Moreover, these genomic regulators can serve as phenotypic switches for GBM cells, as they can affect stemness, proliferation, invasion and DDR. Thus, aberrant expression of such transcripts may facilitate therapy resistance and responsiveness, leading to tumour recurrence (Refs Reference Stackhouse26, Reference Yadav108, Reference Peng and Croce128, Reference DeSouza147).
Gene-silencing technique was employed by Li et al. to demonstrate that silencing lncRNA SNHG15 had a beneficial outcome leading to suppression of GBM tumorigenesis, while also restoring TMZ sensitivity in vitro (Ref. Reference Li148). In 2022, Xu et al. conducted an innovative approach on the lncRNA PRADX. PRADX overexpression activates STAT3 phosphorylation and enhances ACSL1 expression, being associated with accelerated cellular metabolism and tumour growth. Combined ACSL1 and CPT1 inhibitors could reverse this malignant phenotype, which provides the means to further explore lncRNA PRADX as a potential therapeutic target (Ref. Reference Xu149).
HMMR-AS1 was found upregulated following radiation therapy along with the increased expression of DDR proteins ATM, RAD51 and BMI1. Collectively, these findings confirm that chemo- and radiation-induced DDR could activate lncRNAs in GBM, making them attractive as potential therapeutic targets (Refs Reference Stackhouse26, Reference Li150, Reference Yuan151).
Zhang et al. reported that the overexpression of SBF2-AS1 backs the chemoresistant phenotype behind the TMZ-resistant GBM cells. Their study reported that SBF2-AS1 functions as a ceRNA for miR-151a-3p, upregulating its endogenous target, XRCC4, which enhances DSB repair in GBM cells. These results showed that lncSBF2-AS1/miR-151a-3p/XRCC4 axis is involved in the DDR-regulation of TMZ resistance in GBM cells (Ref. Reference Zhang152).
MALAT1 is a well-studied lncRNA that is linked with the activation of DDR pathway. Activation of DDR pathway by TMZ induces overexpression of MALAT1 which is linked via NF-кB to p53. Down-regulation of MALAT1 using nanoparticle-encapsulated anti-MALAT1 siRNA were able to restore the chemosensibility to TMZ, making it an attractive target for the chemosensitization of GBM (Ref. Reference Voce153).
A summary of additional overexpressed lncRNAs in GBM, their regulatory effect on DDR effector genes, and the treatment-associated phenotype are presented in Table 3.
Table 3. Overexpressed lncRNAs in GBM-associated DDR and the effect upon treatment

DDR and circRNA in GBM
CircRNAs are a particular group of ncRNAs produced mainly via back-splicing of pre-mRNA (Ref. Reference Hao167). They are most abundant in brain tissues and found to be highly dysregulated in GBM, where they play significant roles in tumour growth, metastasis, epithelial-to-mesenchymal transition and therapy resistance (Refs Reference Rybak-Wolf168, Reference Sun169).
In a recent study conducted on GBM cells, Wang et al. found that low-dose RI could trigger the production of exosomes carrying cargoes abundant in circ-METRN, which in turn led to increased levels of γH2AX. Thus, circ-METRN was reported to exhibit oncogenic functions, such as GBM progression and radioresistance, by deregulation of DDR-associated γH2AX via miR-4709-3p/GRB14/PDGFRα pathway (Ref. Reference Wang170).
Lou et al. found CDR1as is a particularly interesting circRNA as its expression decreases with the increase of glioma grade, which promotes it as a reliable predictor for overall survival, especially in GBM. The researchers reported that CDR1 interacts with the p53 DBD domain, thus disrupting the p53/MDM2 complex formation. This interaction with the p53 protein is essential for maintaining function and protect from additional DNA damage (Ref. Reference Lou171).
Nonetheless, in a circRNAs expression profiling study conducted by Wang et al. on GBM patients, it was found that compared with the adjacent normal brain tissues, 254 circRNAs were upregulated and 361 circRNAs were downregulated in IDH-wt GBM. In fact, a comprehensive Gene Ontology analysis conducted by the same research group indicated that these differentially expressed circRNAs could be involved in different GBM-associated processes, including DDR and repair (Refs Reference Ferri12, Reference Wang172).
The expression level of circHEATR5B is generally low in tissues and cells, being involved in aerobic glycolysis, a metabolic hallmark of GBM. However, Song et al. reported that circHEATR5B transfection-based overexpression contributed to suppressing the aerobic glycolysis process and GBM cells proliferation in vitro. Moreover, circHEATR5B overexpression proved to play a role in the inhibition of GBM xenograft growth, while also prolonging the survival rate of nude mice. This highlights the potential use of circHEATR5B for the advance of anti-GBM targeted therapies (Ref. Reference Song173). Similarly, Jiang et al. found that circLRFN5 is downregulated in GBM and associated with poor patient prognosis (Ref. Reference Jiang174).
Current therapeutic approaches targeting DDR in GBM
As previously mentioned, DDR pathway is activated by SSB or DSB which are induced by RT or TMZ. Using this pathway, GBM cells acquired resistance to genotoxic anti-tumoral agents. PARP-1 plays a central role in both SSB and DSB, being highly sensitive to detect the DNA damage and favours its repair (Refs Reference Caldecott17, Reference Caron175, Reference Pascal176). PARP-1 inhibitors sensitise GBM to RT and chemotherapy (Refs Reference Sim177–Reference Gourley179). The OLA-TMZ-RTE-01 trial (Ref. Reference Kazlauskas180) included 79 participants: 30 in phase I and 49 in phase IIa, participants with unresectable or partially resecable GBM tumours, aged between 18 and 70 years old. The study highlighted the benefits of PARP-1 inhibitor Olaparib, alongside RT and TMZ, at improving the 18 months' overall survival in patients with unresectable or partially resecable GBM, without harming the non-cancerous brain tissue and without affecting patients' cognition. The measurement of GBM penetration of Olaparib, as well as its safety and efficacy associated with TMZ was tested in a study that included 48 patients with recurrent GBM. Olaparib was well represented at the core of GBM as well as the margins and the patients receiving this treatment tolerated it well (Ref. Reference Hanna181).
The VERTU study (Ref. Reference Sim182) included 125 patients (84 in the experiment group and 41 in the standard group) newly diagnosed with MGMT-unmethylated GBM. The experiment group received veliparib 200 mg twice a day and radiation for 6 weeks and veliparib and 40 mg BD and TMZ, while the standard group received just TMZ and RT. Veliparib was well tolerated but the study did not reach statistical significance (Ref. Reference Sim182).
ATR plays an essential role in most replicating cells' survival (Ref. Reference Cimprich and Cortez183). Thus, there are some limitations regarding the treatment with ATR inhibitors, as they could harm both cancerous and noncancerous cells. At the moment, there are no specific ATR inhibitors clinical trials for GBM, due to their increased toxicity in preclinical studies (Refs Reference Bonm and Kesari16, Reference Majd95). Yet, there are ongoing clinical trials that test ATR inhibitors in combination with RT or other chemotherapy: Elimusertib (BAY1895344) with Pembrolizumab for advanced solid tumours (NCT04095273) or Elimusertib with Pembrolizumab and Stereotactic Body Radiation Therapy for recurrent head and neck cancer (NCT04576091) or AZD6738 with Olaparib (AZD2281) for Clear Cell Renal Cell Carcinoma and Advanced Pancreatic Cancer (NCT03682289).
ATM inhibitors are a practical solution to resistant GBM since they enhance the toxic effects of RT and chemotherapy (Ref. Reference Frosina184). AZD1390 is one of the latest, highly effective ATM inhibitors, being able to effectively cross the blood-brain barrier, in comparison with older-generation ATM inhibitors such as KU-60019 (Refs Reference Jucaite185, Reference Vecchio186). Now, a phase 1 clinical trial is conducted in which AZD1390 in combination with RT being tested on 120 patients with primary/recurrent GBM (NCT03423628).
XPO1 is a protein transporter that facilitates the exports of proteins from the nucleus. XPO1 is upregulated in GBM and other cancers, thus being a potential effective antitumoral target (Ref. Reference Green187). The only XPO1 inhibitors available are Selinexor and Eltanexor. The first is currently given only in haematological malignancies, such as relapsed or refractory multiple myeloma or diffuse large B-cell lymphoma, while the latter is still undergoing clinical trials. (Ref. Reference Martin92). In vivo and in vitro studies showed that Selinexor has radiosensitizing effects against GBM; in addition, it also affects gene translation, since XPO1 also facilitates the transport of ribosomal RNA across the nuclear membrane (Ref. Reference Degorre188). The KING trial (NCT01986348) was a phase II study using Selinexor conducted with 76 participants divided into 4 arms with various treatment regimens. Only at 80 mg/week Selinexor induced responses and had a relevant 6-month progression-free survival rate. Although there were some haematological adverse effects (thrombocytopenia, neutropenia and anaemia) they were reversible with the adjustment of dose (Ref. Reference Lassman189). At the moment, Selinexor is tested in phase 1 clinical trial with 68 participants with recurrent and refractory paediatric solid tumours, including CNS tumours and GBM (NCT02323880).
Wee1 is a protein kinase of the ATR-CHK1 pathway. Its key role is to lengthen the G2 cellular phase, thus making it possible for DDR mechanisms to repair the injured DNA. One Wee1 activator is phosphatidylinositol 3-kinase (PI3 K) inhibition, as an adaptative mechanism of GBM cells (Ref. Reference Wu190). Moreover, it was demonstrated a beneficial association between WEE1 inhibitors and PI3 K inhibitors in GBM therapy (Ref. Reference Wu190). A recent phase 0 clinical trial with 20 participants (NCT02207010) highlighted only that WEE1 inhibitor Adavosertib (AZD1775) passes through the blood-brain barrier and reaches the, and not its efficiency in fighting the tumour (Ref. Reference Sanai191). The 20 participants were grouped into three cohorts and received a single dose of 100, 200 or 400 mg before tumour resection. Part 1 of the study planned the tumoral resection 8 hours post-AZD1775 administration in each cohort; in contrast, in part 2 the resection was planned for 8 hours or 24 hours. In the case of Adavosertib resistance, the biomarker Myt1 should be investigated, because it demonstrated an upregulation of Myt1 following WEE1 inhibitors treatment (Ref. Reference Lewis192).
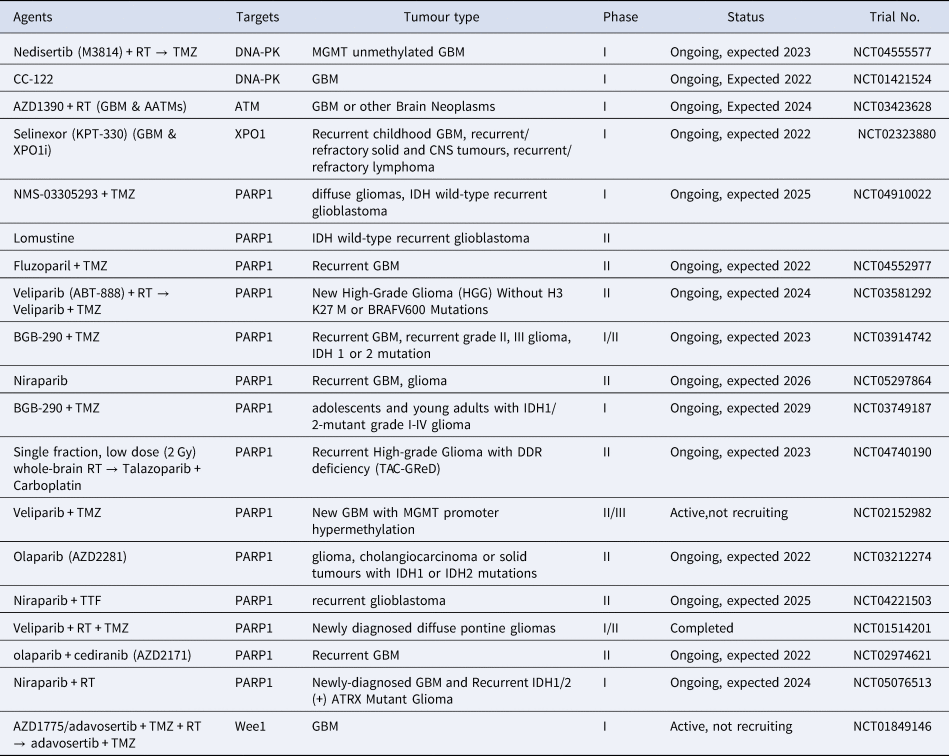
Currently, there are no clinical trials that involve ncRNAs that target DDR pathway in GBM or are being used for disease monitoring. However, in the future these molecules could become of interest as we showed that they are important key regulators of DDR pathway and can be either used as single targets or as adjuvant therapy to current approaches Table 4.
Table 4. Current ongoing clinical trials with various DDR targets
Conclusion
In conclusion, we consider that an in-depth characterisation of the molecular mechanisms involved in DDR can provide important insights into this particular field of GBM biology that can be exploited by the upcoming new DDR inhibitors. An integrated approach needs to consider the underlying genomic background of each individual GBM patient, to check for DDR pathway status in the tumour using both ncRNAs and protein biomarkers and to identify the genomic vulnerability that can be targeted in the particular genomic context of the tumour. By targeting specific vulnerable targets of the DDR pathway using above-mentioned, inhibitors we can try to overcome the current challenges in chemotherapy and RT resistance.
The novelty of this review resides in including in the regulatory loop of DDR in glioblastoma the roles of ncRNAs with a special focus on miRNAs and lncRNAs. We consider that an integrative view over the DDR pathway in glioblastoma which considers ncRNAs can fill the gaps in understanding that limited more consistent progression in this field. NcRNAs can be used assess the functionality of the DDR mechanism and to assess in dynamic treatment response with DDR inhibitors such as PARPi. The widespread distribution of ncRNAs, stability and sensibility are important characteristics that make them attractive biomarker for identifying and monitoring GBM patients in further clinical trials.
Financial support
This paper was supported by the following projects: Clinical and economical impact of personalised targeted anti-microRNA therapies in reconverting lung cancer chemoresistance-CANTEMIR, grant no. 35/01.09.2016; MySMIS 103375, PDI-PFECDI 2021, entitled Increasing the Performance of Scientific Research, Supporting Excellence in Medical Research and Innovation, PROGRES, no. 40PFE/30.12.2021 and SEE 21-COP-0049: Strategic inter-university cooperation to improve research abilities for Ph.D. students for higher educational quality, Excellence in research and development of non-coding RNA DIAGnostics in Oncology -RNADIAGON H2020-MSCARISE-2018- GA no. 824036. The first author, R.P., received an internal grant for Ph.D. students offered by The ‘Iuliu Hatieganu’ University of Medicine and Pharmacy Cluj-Napoca, Romania no. 1529/54 18.01.2019
Conflicts of interest
The authors declare no conflict of interest.






