



Introduction
Deep-sea hydrothermal vent ecosystems are unique and extreme among marine environments, characterized by high pressure, high temperature (up to 390°C), low oxygen and high levels of toxins (hydrogen sulphide, methane and various heavy metals) (Van Dover, Reference Van Dover2000). In such harsh environments, however, there exists lush biological community sustained by chemosynthetic primary production from free-living and symbiotic microbes (Dubilier et al., Reference Dubilier, Bergin and Lott2008).
The shrimp Shinkaicaris leurokolos Kikuchi and Hashimoto, 2000, is one of the representative species of the Okinawa Trough hydrothermal vent area in the Northwest Pacific Ocean (Watanabe and Kojima, Reference Watanabe, Kojima, Ishibashi, Okino and Sunamura2015). This species is specifically distributed in the area very close to the vent that can even contact the hydrothermal fluid instantaneously (Yahagi et al., Reference Yahagi, Watanabe, Ishibashi and Kojima2015), which is expected to have high thermal resistance and anti-chemical toxicity ability. It offers a biological model for uncovering the mechanisms of animals’ adaptation to extreme deep-sea hydrothermal vent environments. Genomic data, especially whole genome map, are essential for clarifying this issue at molecular level.
The genomes of decapods are challenging to assemble due to their large size and complexity (Yuan et al., Reference Yuan, Gao, Zhang, Wei, Liu, Li and Xiang2017). Thus far, no whole-genome map of deep-sea decapods has been reported. For S. leurokolos, only mitochondrial genome and transcriptome have been sequenced and assembled in order to study the origin, evolution and adaptation of this species (Sun et al., Reference Sun, Hui, Wang and Sha2018a; Wang et al., Reference Wang, Sha and Hui2022a). The lack of genetic and genomic data on S. leurokolos greatly restricts the decipherment of its adaptation to extreme environments. Therefore, it highlights the importance of obtaining the whole-genome sequence of this typical vent shrimp, and before this, knowledge of genome size and characteristics is a necessary prerequisite.
Genome survey sequencing (GSS) using next-generation sequencing is currently an important and cost-effective approach to evaluate genome information such as genome size, GC content, heterozygosity and repeat content, as well as developing molecular markers (Li et al., Reference Li, Song, Jin, Li, Gong and Wang2019; Baeza, Reference Baeza2020, Reference Baeza2021; Baeza et al., Reference Baeza, Baker and Liu2022; Choi et al., Reference Choi, Kim, Lee, Jo, Kim, Kim, Parker, Chi and Park2021). In the present study, we aimed to estimate the genomic characteristics of S. leurokolos through GSS, identify repetitive elements in the nuclear genome and assemble a complete mitochondrial genome. These data are expected to provide basic information on the S. leurokolos genome and serve as a framework for subsequent whole-genome map construction.
Materials and methods
Sample collection
Shrimps of S. leurokolos (Figure 1) were collected at Iheya North hydrothermal vent in the Okinawa Trough (126°53.80’E, 27°47.46’N, depth 970 m) during the cruise by the scientific research vessel (RV) KEXUE in July 2018. Species-level morphological identification abided by the main points of Komai and Segonzac (Reference Komai and Segonzac2005). Once aboard, specimens were immediately frozen in liquid nitrogen and stored at −80°C until DNA extraction. One specimen of S. leurokolos was subsequently subjected to genome sequencing.

Figure 1. Swarms of S. leurokolos individuals (marked in the red circle) are crowded along the hydrothermal vent of Iheya North.
DNA extraction, library construction and sequencing
Total genomic DNA was extracted from the muscle tissue using a DNeasy tissue kit (Qiagen, Beijing, China) according to the manufacturer's protocol. The quality and purity of the DNA were detected with NanoDrop and 1% agarose gel electrophoresis. After DNA extraction and detection, high-quality DNA was fragmented using ultrasonic crusher. The sequencing library with an insert size 300–350 bp was constructed with VAHTS Universal DNA Library Prep Kit for Illumina V3 following the manufacturer's recommendations. Paired-end sequencing was conducted using DNBSEQ-T7 platform (MGI Tech Co., Ltd. in Shenzhen, China) by Wuhan Onemore-tech Co., Ltd.
Sequence quality control and genome assembly
The quality control of raw data was performed using the FastQC v0.11.9 (Andrews, Reference Andrews2010) and Trimmomatic v0.39 (Bolger et al., Reference Bolger, Lohse and Usadel2014) based on the four criteria: (1) removing the A-tail and adaptors, (2) deleting the low-quality reads with N content more than 10%, (3) filtering the reads with base quality less than 10 and (4) discarding duplicated reads. Then the clean data were submitted to the Sequence Read Archive (SRA) databank (http://www.ncbi.nlm.nih.gov/sra/), and were available under the accession number PRJNA926015. Genome size, heterozygosity and repeat content of S. leurokolos were estimated based on a K-mer method by Jellyfish and GenomeScope with parameters of 17-mer, 21-mer, 27-mer and 31-mer (Marçais and Kingsford, Reference Marçais and Kingsford2011; Vurture et al., Reference Vurture, Sedlazeck, Nattestad, Underwood, Fang, Gurtowski and Schatz2017). Based on clean data, the draft genome of S. leurokolos was de novo assembled using SOAPdenovo2 (Luo et al., Reference Luo, Liu, Xie, Li, Huang, Yuan, He, Chen, Pan, Liu, Tang, Wu, Zhang, Shi, Liu, Yu, Wang, Lu, Han, Cheung, Yiu, Peng, Zhu, Liu, Liao, Li, Yang, Wang, Lam and Wang2012) with K-mer = 41 and K-mer = 63.
Genomic repetitive elements and microsatellite identification
In the present study, two methods were used for the discovery, annotation and quantification of the repetitive elements from the draft genome of S. leurokolos. First, repetitive elements were de novo annotated using the RepeatModeler v2.0.3 (Flynn et al., Reference Flynn, Hubley, Goubert, Rosen, Clark, Feschotte and Smit2020) and LTR_FINDER v1.0.2 (Xu and Wang, Reference Xu and Wang2007). Second, repetitive sequences were identified by RepeatMasker v4.0.9 (Tempel, Reference Tempel and Bigot2012) and RepeatProteinMask v4.1.0 (a component of the RepeatMasker application) with the Repbase database. The Perl script MISA (http://pgrc.ipk-gatersleben.de/misa/misa.html) was used to identify SSRs in the draft genome of S. leurokolos, and search parameters were set as minimum of 6, 5, 5, 5 and 5 repeats for detecting di-, tri-, tetra-, penta- and hexanucleotide motifs, respectively.
Mitochondrial genome assembly and SNP identification
The mitochondrial genome of S. leurokolos was de novo assembled with Novoplasty v.4.3.1 (Dierckxsens et al., Reference Dierckxsens, Mardulyn and Smits2016) using the published COI sequence of S. leurokolos (GenBank accession no. MH398102) as seed sequence. GapCloser v1.12 was used to fill in the missing regions to acquire the complete circular mitochondrial genome. The mitochondrial genome was annotated using the automatic annotators of mitochondrial genes online, Geseq (Tillich et al., Reference Tillich, Lehwark, Pellizzer, Ulbricht-Jones, Fischer, Bock and Greiner2017) and the MITOS 2 Web server with the invertebrate genetic codes (Donath et al., Reference Donath, Jühling, Al-Arab, Bernhart, Reinhardt, Stadler, Middendorf and Bernt2019), followed by strictly manual check.
To identify variation in S. leurokolos mitochondrial genome, single nucleotide polymorphisms (SNPs) recovery was performed. The previously published S. leurokolos mitochondrial genome (GenBank accession no. MF627741) was set as a reference. Alignment between the two mitochondrial genome sequences was performed using the software MEGA v7.00 (Kumar et al., Reference Kumar, Stecher and Tamura2016). The varied sites were supposed to be candidate SNP markers.
Results and discussion
Sequencing and quality evaluation
A total of 639.75 Gb raw reads were generated for S. leurokolos. After filtering and correction, a total of 599.63 Gb clean reads were derived (Table 1). The Q20 and Q30 values of the sequencing data were 96.28 and 91.18%, respectively (Table 1). It has been specified that Q20 and Q30 values should be at least 90 and 85% (Li et al., Reference Li, Song, Jin, Li, Gong and Wang2019). Therefore, the sequencing data of S. leurokolos genome show extreme precision in the present study. GC content is an important factor in many experiments and bioinformatic analysis, especially for next-generation sequencing where the sequenced DNA has gone through multiple rounds of PCR amplification. High or low GC content will reduce sequencing coverage and cause sequencing bias (Bentley et al., Reference Bentley, Balasubramanian, Swerdlow, Smith, Milton, Brown, Hall, Evers, Barnes and Bignell2008; Aird et al., Reference Aird, Ross, Chen, Danielsson, Fennell, Russ, Jaffe, Nusbaum and Gnirke2011; Cheung et al., Reference Cheung, Down, Latorre and Ahringer2011). In this study, GC content of S. leurokolos sequences was 37.6% showing a mid GC content (30–47%) (Shangguan et al., Reference Shangguan, Han, Kayesh, Sun, Zhang, Pervaiz, Wen and Fang2013). Overall, these results indicate high-quality sequencing data obtained for S. leurokolos.
Table 1. Summary information for the S. leurokolos genome sequencing and genome assembly

Q20: the ratio of data with accuracy above 99% in total data. Q30: the ratio of data with accuracy above 99.90% in total data
K-mer analysis and genome size estimation
The genome size, heterozygosity and repetitive ratio of S. leurokolos were evaluated using K-mer distribution analysis, and the 17-mer yielded the highest model fit (Figure 2 and Table 2). K-mer analysis revealed a unique bimodal profile with a high heterozygous peak around 50× coverage and a lower homozygous peak around 100× coverage (Figure 2). By calculation, the genome size of S. leurokolos was estimated to be 5.08 Gb (Table 2). Flow cytometry is another method for the prediction of genome size. Previous study for other four alvinocaridid shrimps based on flow cytometry reveals that genome sizes range from 10,160 Mp in Rimicaris exoculata to 13,050 Mp in Chorocaris chacei (Bonnivard et al., Reference Bonnivard, Catrice, Ravaux, Brown and Higuet2009), displaying a large genome size in the family Alvinocarididae. It seems that the genome size of S. leurokolos is much smaller than those of other alvinocaridid shrimps, or its genome size has been underestimated by GSS. The significant discordance of genome size revealed by GSS and flow cytometry has been also detected in other decapods, such as crayfish Procambarus clarkia, showing larger genome size by flow cytometry analysis than that revealed by GSS (Shi et al., Reference Shi, Yi and Li2018). However, muscle instead of haemolymph cell has been used in the flow cytometry analysis for alvinocaridid shrimps (Bonnivard et al., Reference Bonnivard, Catrice, Ravaux, Brown and Higuet2009), which might be due to the difficulty in collecting living shrimp samples from deep sea. It may influence the quality of cell suspension preparation and in turn affect the precision of genome size estimation. On the other hand, the high heterozygosity and repeat ratio characteristics of S. leurokolos genome as shown below might bring biased results in genome size estimation by affecting the K-mer depth distribution (Shi et al., Reference Shi, Yi and Li2018). In brief, GSS and flow cytometry should be combined to estimate genome sizes of deep-sea species with large and complex genome, and the genome size of S. leurokolos might be larger than 5.08 Gb.
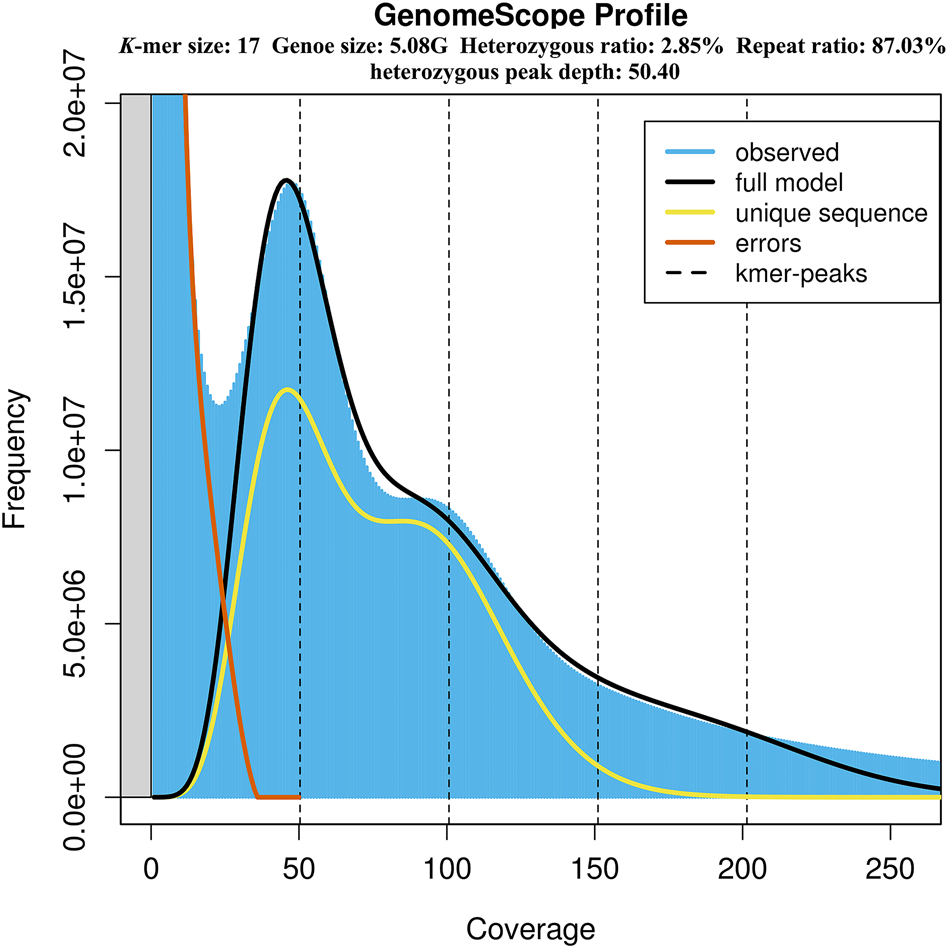
Figure 2. K-mer (K = 17) analysis for estimation of the genome size of S. leurokolos. The x-axis represents coverage, and the y-axis represents the frequency at each depth.
Table 2. Statistics of the estimated S. leurokolos genome size and other characteristics

According to the K-mer distribution, an extremely high heterozygosity 2.85% was detected in S. leurokolos genome (Figure 2 and Table 2). It has been suggested that genome assembly will be difficult if the heterozygosity rate exceeds 0.5%, and it is even more difficult if it exceeds 1% (Marçais and Kingsford, Reference Marçais and Kingsford2011). The repeat ratio of S. leurokolos genomic sequences was also high (87.03%) (Figure 2 and Table 2). The high heterozygosity rate and repeat ratio have been also revealed in other decapods, such as Litopenaeus vannamei, Penaeus chinensis and P. monodon (Zhang et al., Reference Zhang, Yuan, Sun, Li, Gao, Yu, Liu, Wang, Lv, Zhang, Ma, Wang, Lin, Wang, Zhu, Zhang, Zhang, Jin, Yu, Kong, Xu, Chen, Zhang, Sorgeloos, Sagi, Alcivar-Warren, Liu, Wang, Ruan, Chu, Liu, Li and Xiang2019; Van Quyen et al., Reference Van Quyen, Gan, Lee, Nguyen, Nguyen, Tran, Nguyen, Khang and Austin2020; Uengwetwanit et al., Reference Uengwetwanit, Pootakham, Nookaew, Sonthirod, Angthong, Sittikankaew, Rungrassamee, Arayamethakorn, Wongsurawat, Jenjaroenpun, Sangsrakru, Leelatanawit, Khudet, Koehorst, Schaap, Martins dos Santos, Tangy and Karoonuthaisiri2021; Yuan et al., Reference Yuan, Zhang, Wang, Sun, Liu, Li, Yu, Gao, Liu, Zhang, Kong, Fan, Zhang, Feng, Xiang and Li2021b; Wang et al., Reference Wang, Ren, Liu, Li, Lv, Wang, Zhang, Wei, Zhou, He and Li2022b), and difficulties in genome assembly seem to be common problem in decapods due to high heterozygosity and repeat ratio (Yuan et al., Reference Yuan, Zhang, Li and Xiang2021a).
Genome de novo assembly
To assemble the draft genome of S. leurokolos, two K-mer values, 41 and 63 bp were selected. Unfortunately, too much computer memory was required and the assembly task could not be completed when using the 41 bp K-mer value. A complete assembly using 63 bp K-mer value was obtained (Table 1). Finally, our efforts recovered a total of 9,527,856,577 bp scaffolds with the scaffold N50 value of 597 bp, and the maximum scaffold was 69,344 bp in length (Table 1). It is apparent that the size of draft genome assembly is almost twice as large as the estimated genome size based on 17-mer analysis. The most plausible explanation for the genome assembly size deviation may be that the presence of a large number of repetitive elements (87.03%) and high heterozygosity (2.85%) of S. leurokolo genome might induce the assembly has multiple copies of the same genomic region and even contained two divergent haplotypes (Pflug et al., Reference Pflug, Holmes, Burrus, Johnston and Maddison2020; Hu et al., Reference Hu, Feng, Xiang, Wang, Salojärvi, Liu, Wu, Zhang, Liang, Jiang, Liu, Ou, Li, Fan, Mai, Chen, Zhang, Zheng, Zhang, Peng, Yao, Wai, Luo, Fu J, Tang, Lan, Lai, Sun, Wei, Li, Chen, Huang, Yan, Liu, McHale, Rolling, Guyot, Sankoff, Zheng, Albert, Ming, Chen, Xia and Li2022; Wyngaard et al., Reference Wyngaard, Skern-Mauritzen, Malde, Prendergast and Peruzzi2022). The average GC content of S. leurokolos assembled genome was about 36.12%. To further evaluate the data of our assembly, we compared it to previously reported genome survey data of decapods. The scaffold N50 of S. leurokolos is much shorter than that of Pacific white shrimp L. vannamei (1343 bp) (Yu et al., Reference Yu, Zhang, Yuan, Li, Chen, Zhao, Huang, Zheng and Xiang2015) and red swamp crayfish P. clarkia (1426 bp) (Shi et al., Reference Shi, Yi and Li2018). The inherent defects of second-generation sequencing technology in read length and high complexity of the large genome of S. leurokolos itself should be the main reasons for the poor assembly. We hold the opinion that the large and complex genome of S. leurokolos represents typical challenges faced by all alvinocaridid shrimp genomes, which partly explains why genomic resources for alvinocaridid shrimps are so limited compared to those of many other deep-sea organisms. Hence, developing new assemblers and bioinformatics tools and using combination of short- and long-read sequencing technologies (i.e. PacBio, Oxford Nanopore Technologies, ONT) are expected to solve these challenges for assembling a high-quality genome. The current GSS data could serve as a reference for subsequent whole-genome sequencing project of S. leurokolos.
Genomic repetitive elements annotation
Repetitive sequences, especially transposable elements (TEs), are known to be an evolutionary precursor of many genes, a driving force in the evolution of epigenetic regulation and an important factor in genomic stability maintenance and evolution (Jurka et al., Reference Jurka, Kapitonov, Kohany and Jurka2007). In total, 4250 Mb repetitive elements were identified in S. leurokolos draft genome, accounting for 44.62% of the assembled genome (Table 3). Combining the results from RepeatMasker and RepeatProteinMask analyses, our results revealed that among these repetitive sequences, 38.92% (3708 Mb) were TEs, but 16.49% could not be classified within the TEs (Table 4). Long interspersed nuclear elements (LINEs) were the most common among the TEs, accounting for 10.45%, followed by DNA transposons (6.09%) and long-terminal repeat elements (LTRs) (4.79%) (Table 4). These repetitive elements, including LINEs, DNA and LTRs, also take up a large proportion of genomes in many other decapod crustaceans (Baeza, Reference Baeza2020; Tang et al., Reference Tang, Wang, Liu, Zhang, Jiang, Li, Wang, Sun, Sha, Jiang, Wu, Ren, Li, Xuan, Ge, Jiang, She, Sun, Qiu, Wang, Wang, Qiu, Zhang and Li2020; Chak et al., Reference Chak, Harris, Hultgren, Jeffery and Rubenstein2021; Uengwetwanit et al., Reference Uengwetwanit, Pootakham, Nookaew, Sonthirod, Angthong, Sittikankaew, Rungrassamee, Arayamethakorn, Wongsurawat, Jenjaroenpun, Sangsrakru, Leelatanawit, Khudet, Koehorst, Schaap, Martins dos Santos, Tangy and Karoonuthaisiri2021). However, it has been suggested that the ‘unclassified’ TEs with a large proportion may contain species-specific variants of known repetitive elements, and we should be cautious when comparing these datasets directly with those of other species (Murgarella et al., Reference Murgarella, Puiu, Novoa, Figueras, Posada and Canchaya2016).
Table 3. Statistics of repetitive sequence annotation in the S. leurokolos draft genome assembly

Table 4. Statistics of TEs in the S. leurokolos draft genome assembly

RepBase TEs and TE proteins were obtained based on the RepBase library using RepeatMasker and RepeatProteinMask, respectively. De novo repeat prediction was performed using RepeatMasker against the de novo repeat library of S. leurokolos, which was constructed by the programs LTR_FINDER and RepeatModeler. Combined TEs were the union of the three methods.
Microsatellite analysis
It is widely recognized that as a most popular and versatile genetic marker, SSRs are widely used for the genetic characterization of populations due to their abundance in genome, high polymorphism and co-dominant nature (Abdul-Muneer, Reference Abdul-Muneer2014). In the assembled scaffolds, a total of 12,121,553 microsatellite motifs were identified in S. leurokolos (Table 5). Among them, the di-nucleotide was the most abundant, accounting for 70.27% of the total SSRs, which was followed by tri- (25.54%), tetra- (3.33%), penta- (0.50%) and hexa- (3.36%) nucleotide SSRs (Table 6). Our finding shows that both di-nucleotide and tri-nucleotide SSRs are numerous, and the number of repetitions is inversely proportional to the length of repetitions. This result is consistent with those in other crustaceans, such as kuruma prawn Marsupenaeus japonicus (Lu et al., Reference Lu, Luan, Kong, Hu, Mao and Zhong2017), Japanese mantis shrimp Oratosquilla oratoria (Cheng et al., Reference Cheng, Zhang and Sha2018) and Antarctic krill Euphausia superba (Huang et al., Reference Huang, Bian, Liu, Wang, Xue, Huang, Yi, You, Song, Mao, Song and Shi2020). It has been proposed that longer repeats have downward mutation bias and short persistence times (Harr and Schlötterer, Reference Harr and Schlötterer2000), and therefore, less SSRs with longer repeat units exist in genomes.
Table 5. Statistics of SSR distribution in the S. leurokolos draft genome assembly

Table 6. Organization of the S. leurokolos mitogenome

Mitochondrial genome and candidate molecular marker identification
Mitochondria are essential organelles that generate most chemical energy to power the cell's biochemical reactions. There is evidence that mitochondrial DNA plays a role in many aspects of biological life history, such as lifespan, fertility, resistance to starvation, altitude adaptation and regulation of temperature (Ballard and Melvin, Reference Ballard and Melvin2010). It is therefore of significant importance to investigate the mitochondrial genome of S. leurokolos inhabiting deep-sea chemosynthetic ecosystems. In this study, we assembled a 15,906 bp long complete mitochondrial genome (GenBank accession no. OQ622002) of S. leurokolos from the GSS data. It consisted of 13 protein-coding genes (PCGs), 2 ribosomal RNA genes (rrnS and rrnL), 22 transfer (tRNA) genes and a non-coding hypervariable control region (1026 bp) between rrnS and tRNA-Ile, showing the typical alvinocaridid shrimp mitogenome arrangement model (Table 6). Most of the PCGs and tRNA genes were encoded on the positive strand. Gene overlaps in 19 gene junctions (a total of 57 bp in length) and intergenic spaces in 14 gene junctions (ranging from 1 to 50 bp) were also observed (Table 6).
Moreover, mitochondrial DNA fragments have been proved to be efficient molecular markers in phylogenetic and population genetic analysis. In order to identify candidate markers, we aligned the mitochondrial genome assembled in this study with the previous reported S. leurokolos mitochondrial genome (Sun et al., Reference Sun, Hui, Wang and Sha2018a). By comparison, 3 indels (all located in the control region) and 71 SNPs were detected. The SNPs included 66 transitions and 5 transversions: 47 in PCGs, 3 in tRNAs, 1 in rRNAs and 19 in non-coding regions. Of the 47 SNPs in PCGs, only four mutations were non-synonymous substitutions (Table 7), which occurred in cox1, nad2, cytb and nad1 (Table 7). It is a general observation in molecular evolution that functional importance and substitution rate are negatively correlated (Sun et al., Reference Sun, Li and Kong2010). This means that the more functionally important genes (or genetic regions) evolve more slowly due to their important effects or strong functional constraints (Kimura, Reference Kimura1983; Yang, Reference Yang2006). In addition, the relatively high substitution rates observed in tRNA-Ala (1.59%), control region (1.58%), tRNA-Cys (1.49%) and tRNA-Trp (1.33%) may indicate relatively low functional constraints in these regions.
Table 7. Summary of SNPs in S. leurokolos mitochondrial genome

To date, population genetic and phylogenetic studies for alvinocaridid shrimps are mainly based on mitochondrial cox1, 12S rDNA and 16S rDNA genes (Yahagi et al., Reference Yahagi, Watanabe, Ishibashi and Kojima2015; Sun et al., Reference Sun, Sha and Wang2018b). In this study, cox1, nad2, nad4 and control region show high mutation rate, and the sequences are long enough for primer design. Hence, these mitochondrial genes can be selected as candidate markers for population genetic studies for S. leurokolos. However, it requires further validation by amplification and sequencing in more individuals.
Conclusions
In summary, this study developed and surveyed the first reference genome for S. leurokolos, an alvinocaridid shrimp from Iheya North hydrothermal vent. It represents the first genome survey for crustaceans from deep-sea chemosynthetic ecosystem. The results showed that the genome of S. leurokolos was extremely complex, with large genome size, extremely high heterozygosity and repeat ratio. The patterns of genome nuclear repetitive elements were investigated, and a large number of SSRs were detected. The mitochondrial genome of S. leurokolos was also assembled, and candidate molecular markers for population genetic study were proposed. These datasets enrich genetic resources of deep-sea life, and are expected to facilitate further studies on the evolutionary biology of alvinocaridid shrimps, as well as the construction of a high-quality genome map of the deep-sea vent S. leurokolos.
Data
The clean data of the genome survey sequencing were openly available in NCBI SRA databank under the accession number PRJNA926015. The authors confirm that the other data supporting the findings of this study are available within the article.
Acknowledgements
The samples were collected by RV KEXUE. The authors wish to thank the crews for their help during collection of samples.
Author contributions
M. H. and Z. S. formulated the research question and designed the study. M. H. collected the specimen. Q. X. extracted DNA of the specimen. A. W. and M.H. carried out the study, analysed the data, interpreted the findings and wrote the article. J. C. and Z. S. also interpreted the findings and revised the article.
Financial support
This work was funded by the Science and Technology Innovation Project of Laoshan Laboratory (LSKJ202203104), the National Science Foundation for Distinguished Young Scholars (42025603) and the Strategic Priority Research Program of Chinese Academy of Sciences (XDB42000000).
Competing interests
None.
Ethical standards
No regulated invertebrate was involved in this study.









