



The most well-known chromosome 22 anomaly associated with CHD is the chromosome 22q11.2 microdeletion, the most common cause of DiGeorge, velocardiofacial, conotruncal anomaly face syndrome, Cayler cardiofacial syndrome, and a subset of patients with Opitz G/BBB syndrome. There are multiple non-cardiac features of this condition including immunodeficiency with absent or hypoplastic thymus, hypoparathyroidism, palatal and velopharyngeal insufficiency, facial dysmorphism, neurodevelopmental delay of varying degrees, and behavioural/psychiatric differences. Reference McDonald-McGinn, Kirschner and Goldmuntz1 Phenotypically, up to 65% of patients Reference Campbell, Sheppard and Crowley2 and 92% of fetuses Reference Noël, Pelluard and Delezoide3 with Chromosome 22q11 deletions have CHD, most often conotruncal defects. Ventricular septal defects are most common at about 25% of patients, followed by tetralogy of Fallot, interrupted aortic arch, and other aortic arch abnormalities. Truncus arteriosus and pulmonary atresia each represent around 5% of lesions. Reference Campbell, Sheppard and Crowley2 Less than 3% of fetuses Reference Noël, Pelluard and Delezoide3 and 1% of patients Reference McDonald-McGinn, Kirschner and Goldmuntz1,Reference Campbell, Sheppard and Crowley2 have been noted to have single ventricle lesions. Duplications of chromosome 22 are seemingly less common and more variable, however there are some small reports showing associations with CHD including conotruncal defects, interrupted aortic arch, and hypoplastic left heart syndrome. Reference Ensenauer, Adeyinka and Flynn4–Reference Weisfeld-Adams, Edelmann, Gadi and Mehta8
Single ventricle CHD is a condition characterised by hypoplasia of either the right or left ventricle and associated atrioventricular valve. There are several underlying genetic conditions associated with hypoplastic right and left heart syndromes. Review of the literature identified only a few case reports presenting single ventricle CHD and abnormalities of chromosome 22, Reference McMahon, Morgan and Greally9–Reference Taylor, Holtby and Macpherson12 but the incidence is unknown and there is no aggregate data describing this population. In this series, we report our institutional experience with chromosome 22q11 deletions and duplications in single ventricle CHD.
Materials and method
We retrospectively reviewed data from the Children’s Hospital of Philadelphia cardiothoracic surgery, cardiology, and genetics databases for patients who had copy number variants within the chromosome 22q11.2 region and underwent surgery for single ventricle cardiac disease. All patients with a 22q11.2 deletion or duplication who had initial surgery between 1 July, 1984 and 30 June, 2021 were included. Patient data were extracted from these databases and individual medical records. This protocol was approved by the Children’s Hospital of Philadelphia Institutional Review Board with a waiver of consent.
Results
Seventeen patients met inclusion criteria. Patient characteristics are detailed in Table 1. Nine patients were female and 8 were male. None of the patients were born at less than 34 weeks of gestational age. The most common types of CHD were hypoplastic left heart syndrome (n = 8) and small left-sided structures (n = 6). Additional cardiac lesions included pulmonary atresia with intact ventricular septum (n = 1), atrioventricular canal (n = 1), and a single ventricle variant of tetralogy of Fallot (n = 1). The right ventricle was dominant in 14 patients. Vascular anomalies included interrupted aortic arch (n = 7), right-sided aortic arch (n = 2), and anomalous right subclavian artery (n = 4). Fifteen of the patients had ductal dependent physiology at birth requiring prostaglandin therapy.
Table 1. Patient Characteristics
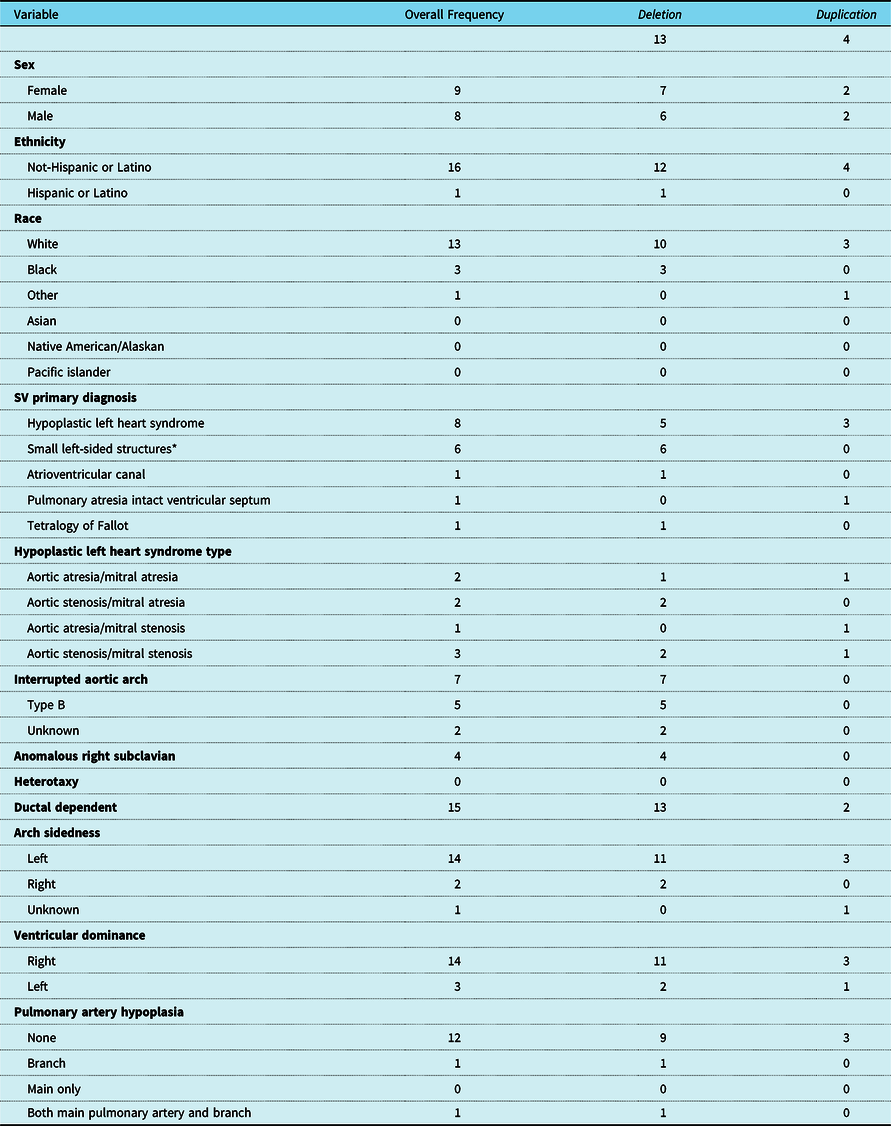
* Includes various combinations of sub aortic stenosis, hypoplastic aortic valve, and hypoplastic left ventricle.
Table 2 details the specifics of the 22q11.2 copy number variants including deletion or duplication size. A Chromosome 22q11.2 deletion was present in 13 patients. The standard microdeletion (low copy repeat 22A through 22D), considered the standard deletion size and inclusive of the important cardiac developmental gene TBX1, was most common in this group (n = 6). Of note, 5 of the 15 patients in the cohort had a confirmed diagnosis of 22q11.2 deletion syndrome by fluorescence in situ hybridisation Reference Newburger, Sleeper and Frommelt13 including the low copy repeat 22A-22B region inclusive of TBX1, but deletion sizing was not available as testing preceded the advent of deletion sizing. In addition, there were four patients with a standard 22q11.2 low copy repeat 22A-22D duplication which has been associated with structural anomalies, including CHD, developmental differences, and autism.
Table 2. Genetic Details

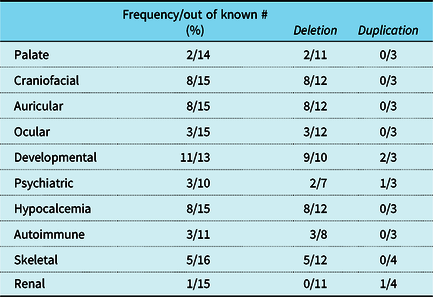
Phenotypic findings typically associated with 22q11.2 deletion syndrome were identified in most patients in this cohort. Craniofacial and auricular abnormalities were documented in 8 patients, but palatal anomalies including an overt cleft palate or velopharyngeal incompetence were seen in only 2 patients. Over half of the patients in the cohort had a history of hypocalcemia and nearly all had documented developmental delay at their most recent follow-up visit. More detail on phenotypic features is provided in Table 3. As has been reported previously, a greater number of structural anomalies were noted in patients with a deletion, but the numbers in our present cohort are too small to determine significance.
Table 3. Presence of 22q Specific Phenotypic Findings
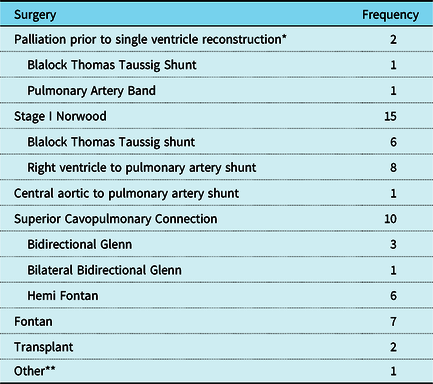
A neonatal intervention was performed in 16 of 17 patients. A Blalock Thomas Taussig shunt was performed on the pulmonary atresia and intact ventricular septum patient who went on to subsequent superior cavopulmonary anastomosis outside of the neonatal period. The tetralogy of Fallot and total anomalous pulmonary venous return patient did not undergo neonatal intervention. A neonatal Stage 1 Norwood operation was performed in 15 patients, one of which had been previously palliated with a pulmonary artery band. Eight patients had a Norwood procedure with a right ventricle to pulmonary conduit, 6 patients had a Norwood procedure with a modified Blalock Thomas Taussig Shunt, and one patient had a Norwood with a central aorta to pulmonary artery shunt.
Superior cavopulmonary anastomosis was performed in 10 patients and 7 patients went on to a Fontan completion. There are 2 patients in the cohort who were alive with a superior cavopulmonary anastomosis awaiting Fontan at follow-up. Two patients underwent cardiac transplantation after full staged reconstruction, at 7 and 13 years after Fontan completion. The patient with tetralogy of Fallot had repair at 8 months of age (atrial septal defect closure, ventricular septal defect closure, transannular right ventricular outflow tract patch, and suture-less repair of total anomalous pulmonary venous return), but subsequently required takedown of the atrial septal defect and ventricular septal defect patches for failure to tolerate 2 ventricle physiology. Details of the operations performed and the patient status at follow-up are shown in Tables 4 and 5. When comparing patients with duplications versus those with deletions, most had hypoplastic left heart syndrome (3 of 4). Of those, one expired after the Norwood operation, one is alive with a Fontan, and one is alive awaiting a Fontan after a superior cavopulmonary anastomosis.
Table 4. Procedures Performed
* The Blalock Thomas Taussig shunt patient went straight to superior cavopulmonary anastomosis and pulmonary artery band patient went on to Stage I.
** Patient initially had sutureless repair of total anomalous pulmonary venous return, tetralogy of Fallot with closure of ventricular septal defect, closure of atrial septal defect, and transannular patch. The patient failed to tolerate the two-ventricle physiology and required takedown of the atrial septal defect and ventricular septal defect patches.
Table 5. Current Status of Individuals in Cohort

* Patient with total anomalous pulmonary venous return and tetralogy of Fallot.
** One patient was alive after superior cavopulmonary anastomosis but has no recent follow-up to know if went on to Fontan.
There were 8 total deaths in the cohort, most of which were hospital mortalities after a Stage I Norwood operation (n = 5). By era, the mortality after a Norwood operation was 67% in the 1980s, 25% in the 1990s, and 20% after 2000. Three of the post-Norwood mortalities were cardiac arrest in the setting of poor ventricular function and one was secondary to dislodgement of an ECMO cannula, and one did not have adequate documentation to determine the cause of death. Four of the 5 post-Norwood mortalities had a diagnosis of hypoplastic left heart syndrome with a restrictive atrial septum and ductal dependent blood flow. Three of the 5 had mild to moderate atrioventricular valve insufficiency on their pre-operative echocardiogram.
The other mortalities included: one patient expired 6 months after the superior cavopulmonary anastomosis operation of septic shock unrelated to surgery, one patient expired of heart failure 11 years after Fontan completion, and the patient with tetralogy of Fallot with total anomalous pulmonary venous return expired at 13 months of age from refractory chronic hypoxemic respiratory failure. Overall survival is 64% at 1 year, and 58% at 5 and 10 years as demonstrated in Fig 1.
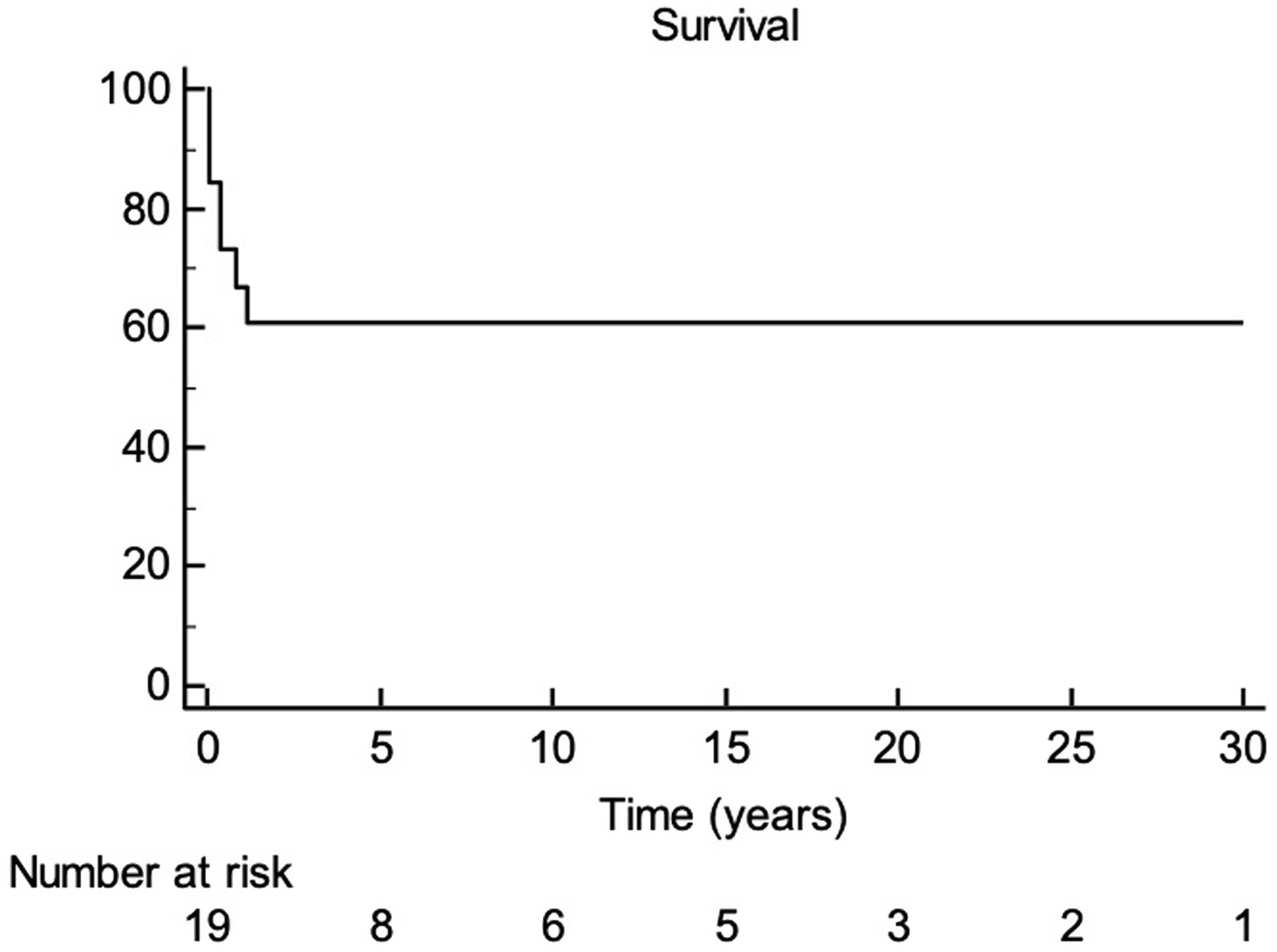
Figure 1 Survival of patients with 22q copy number variants who underwent cardiac surgery for single ventricle disease.
Discussion
Over 36 years, we identified 17 patients with chromosome 22q11.2 copy number variants who underwent surgery for single ventricle CHD. Our data confirms previous reports that this is a relatively rare lesion. Though this cohort is too small to analyse differences in outcomes, we were able to catalogue descriptive data. The most common presentation was a variant of hypoplastic left heart syndrome with the usual 22q11.2 low copy repeat 22A-22D cytogenetic microdeletion. The most common neonatal surgical intervention performed was the Norwood procedure, where most of the mortality burden occurs. Long-term survival is similar to all infants with single ventricle disease, approximately 60% at 6 years of age in the single ventricle reconstruction trial. Reference Newburger, Sleeper and Frommelt13
Deletions of sub-band 11.2 of the long arm of chromosome 22 are estimated to be as common as approximately 1 in 2,000 live births and 1 in 992 pregnancies. Reference Campbell, Sheppard and Crowley2,Reference Grati and Gross14 Though reported less frequently, the 22q11 deletion syndrome has a complementary duplication syndrome in which the same approximately 3 megabase region that is deleted is instead duplicated. This occurs following meiotic non-allelic homologous recombination, due to the presence of low copy repeats, which flank the chromosome 22q11.2 region leading to aberrant inter-chromosomal exchanges resulting in either a duplication or a deletion. Reference Ensenauer, Adeyinka and Flynn4 These deletions cause a range of phenotypes previously classified clinically by separate authors as DiGeorge, velocardiofacial, conotruncal anomaly face syndromes, Cayler cardiofacial syndrome, and a subset of patients with Opitz G/BBB syndrome. The chromosome 22q11.2 microdeletion syndrome manifests with significant phenotypic variability leading to significant morbidity and some mortality, for example: immunodeficiency as a result of absent or hypoplastic thymus, hypocalcaemia/hypoparathyroidism, palatal and velopharyngeal insufficiency, craniofacial dysmorphism, neurodevelopmental delay of varying degrees, psychiatric illness, and CHD. Reference McDonald-McGinn, Kirschner and Goldmuntz1 The most commonly associated types of CHD are tetralogy of Fallot, interrupted aortic arch, and Truncus Arteriosus, with only a small percentage being single ventricle lesions. Reference McDonald-McGinn, Kirschner and Goldmuntz1–Reference Noël, Pelluard and Delezoide3,Reference McDonald-McGinn, Sullivan and Marino15 Some phenotypic findings overlap in patients with deletions and duplications. Patients with duplications can be subject to palate, urogenital abnormalities, hearts defects, hearing loss, cognitive, behavioural defects, and psychiatric abnormalities. However, duplications are associated with hypertelorism, down-slanting palpebral fissures with or without ptosis; and mild micro-/retrognathia; which differs from the typical facies associated with 22q11.2 deletion syndrome where malar flatness, hooded eyelids, auricular and nasal anomalies, and asymmetric crying facies are common. Reference Ensenauer, Adeyinka and Flynn4
Children’s Hospital of Philadelphia has been involved in the clinical care of individuals with chromosome 22q11.2 abnormalities since 1982. We have a continuously growing longitudinal cohort of over 1,700 patients with laboratory confirmed 22q11.2 copy number variant since the introduction of fluorescence in situ hybridisation studies developed at Children’s Hospital of Philadelphia specific to the chromosome 22q11.2 region from 1992 to present. In 2018, our institution published a comprehensive review of patients with 22q11.2 deletion syndrome from our 22q and You Center database. Of over 1400 patients, 64% had CHD. The specific diagnoses included about 25% ventricular septal defects, 20% tetralogy of Fallot, 10% interrupted aortic arch, and about 15% other aortic arch abnormalities. Truncus arteriosus, pulmonary atresia with or without ventricular septal defect each represented around 5% of lesions. Reference Campbell, Sheppard and Crowley2 Additionally, patients had dysfunction or abnormalities of the immune system (77%), palate (67%), gastrointestinal tract (65%), endocrine function (>50%), and vertebrae (50%). Reference Campbell, Sheppard and Crowley2 These patients also had a high incidence of neurologic and psychiatric comorbidities including: attention deficit hyperactivity disorder, autism spectrum disorder, anxiety, behaviour disorders, and psychotic disorders. In addition, significant delays in reaching physical and language developmental milestones were noted. This comprehensive report demonstrates the medical complexity of these patients and emphasises the importance of early diagnosis.
This study on the associations of single ventricle CHD and 22q11.2 copy number variants has some limitations. The data were reviewed retrospectively thus it may be prone to misclassification, selection bias, or confounding. Additionally, we are unable to make clear causal associations between the specific findings that contribute to differences in morbidity or mortality after surgery. The sample size is small, so we are unable to identify if any of the differences seen in this group are statistically significant. Though our centre is a high-volume provider of single ventricle disease and 22q11.2 copy number variants, it is possible that our sample is not representative of the entire population.
Despite the limitations, this study demonstrates that patients with single ventricle CHD often have features associated with a diagnosis of a 22q11.2 copy number variants. As previously mentioned, these phenotypic findings create additional challenges with regards to surgical intervention and cause additional morbidity following cardiac surgery. There are several reports describing longer length of stay and increased morbidity with 22q11 in 2 ventricle CHD, specifically tetralogy of Fallot, truncus arteriosus, and interrupted aortic arch. Reference O’Byrne, Yang and Mercer-Rosa16,Reference Mercer-Rosa, Pinto, Yang, Tanel and Goldmuntz17 Thus, it is important to consider a diagnosis of 22q11.2 copy number variants in patients with single ventricle CHD. The initiation of prompt multidisciplinary care could prove helpful in improving outcomes after cardiac surgical repair in these patients.
Acknowledgements
This work has been made possible by National Institutes of Health funding for Donna McDonald-McGinn and the 22q and You Center including 070454-11, 125757-02, 119737-01, and the Daniel M. Tabas Endowed Chair in Pediatric Cardiac Surgery.
Financial support
This research received no specific grant from any funding agency, commercial, or not-for-profit sectors.
Conflicts of interest
None.
Ethical standards
The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national guidelines and has been approved by Children’s Hospital of Philadelphia Institutional Review Board.
Disclosures
This was presented at the 68th Annual Meeting of the Southern Thoracic Surgical Association.






