


Neuromyelitis optica spectrum disorders (NMOSDs) are a group of autoimmune conditions characterized by inflammatory involvement of the optic nerve, spinal cord (typically longitudinally extensive transverse myelitis or LETM), and the supratentorial central nervous system. The disease typically affects young adults, but improved diagnostic testing with specific autoantibodies against aquaporin 4 (AQP4) has led to increasing rates of diagnosis in older populations.
Late-onset NMOSD (LONMOSD) is usually defined as onset >50 years of age, and it is thought to be more severe than early-onset NMOSD (EONMOSD).Reference Collongues, Marignier and Jacob1 Furthermore, brainstem involvement and area postrema syndrome (APS) are exceedingly rare in LONMOSD. We present the case of a 68-year-old woman who presented with APS and AQP4+ LONMOSD.
A 68-year-old woman presented to the emergency room (ER) with 4 days of nausea, vomiting, and intractable hiccups. Workup for metabolic and infectious causes and a brain computed tomography were negative, and she was referred to the gastroenterology unit for an upper endoscopy that revealed antral gastritis (H. Pylori negative). She was treated with proton pump inhibitors and sucralfate. After 3 weeks, there was no improvement and she developed left arm and leg weakness and presented back to the ER.
On exam, she was alert and cranial nerves were normal (including funduscopy). She had left-sided weakness (4/5) in the upper extremity and in the hip flexors; tone was normal and reflexes were brisk (3+) in the left with upgoing toes. Sensory exam was normal, her gait was unsteady, and she had a positive Romberg test. She denied any recent travel, fevers, or night sweats. Routine laboratory workup was normal, including anti-nuclear antibodies and acute phase reactants. An magnetic resonance imaging (MRI) of the brain and spine revealed multiple non-specific white-matter hyperintensities, subtle changes and enhancement surrounding the 4th ventricle, and an enhancing, longitudinally extensive (C5-T1), left lateral cervical cord lesion (Figure 1). Cerebrospinal fluid testing was bland with a normal protein count, and only five white blood cells. An infectious panel was negative, including herpes simplex virus I and II, varicella zoster virus, and enterovirus. Serum AQP4-antibody was high-positive (cell-based assay), and anti-myelin oligodendrocyte glycoprotein antibodies were negative.
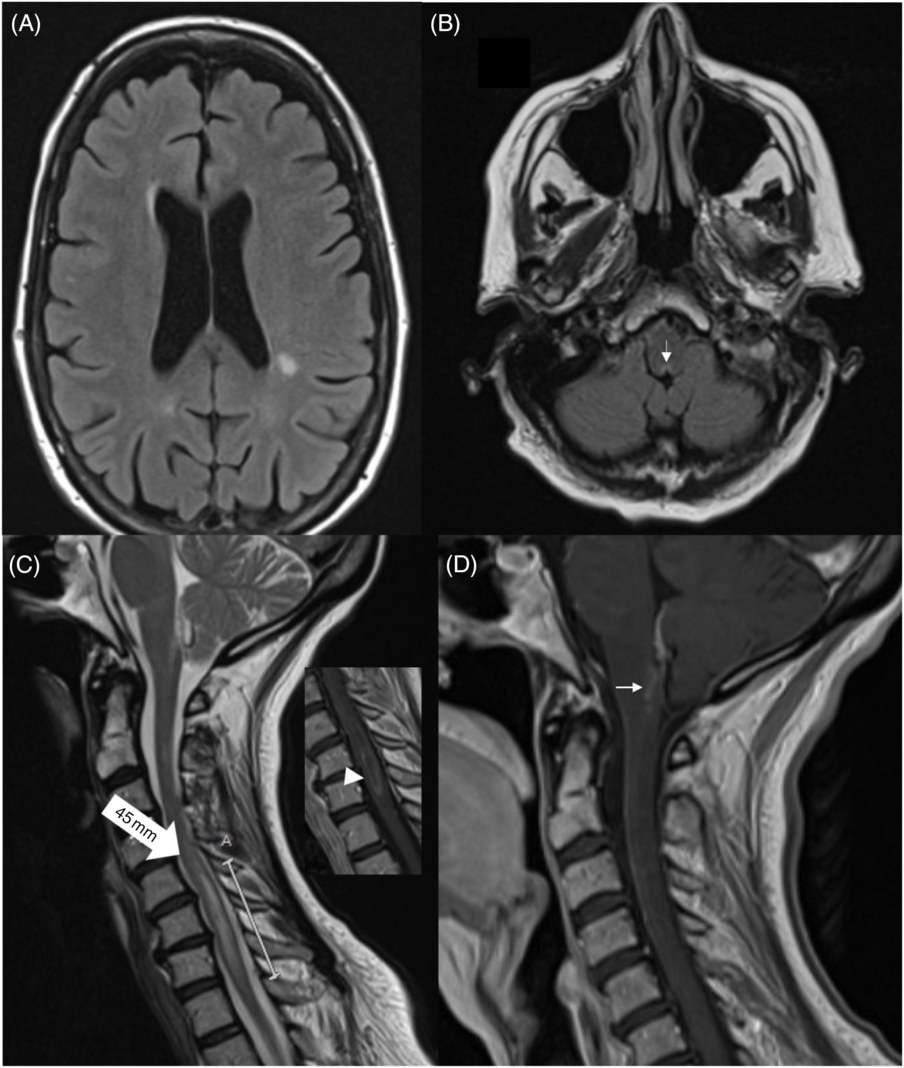
Figure 1: Brain MRI showing (non-enhancing) periventricular white-matter lesions (A). There is a subtle hyperintensity in the posterior medulla, adjacent to the 4th ventricle (B, arrow). Spinal MRI revealed a 45-mm hyperintense lesion in C5-T1 (C, large arrow). The lesion showed subtle enhancement (insert, arrowhead). There was enhancement around the periaqueductal posterior medulla (D).
NMOSD was diagnosed, and she was started on 1 g of IV methylprednisolone. After five doses, she showed no improvement, and she was transitioned to plasmapheresis (PLEX). After five sessions (every other day), her weakness improved and her brainstem symptoms resolved. She was treated with prednisone 50 mg/day and was started on rituximab for maintenance therapy.
APS occurs when there is involvement of the area postrema, a chemosensitive region of the brainstem known to be the “vomiting center” of the brain,Reference Camara-Lemarroy and Burton2 and strongly suggests NMOSD. Patients with LONMOSD have an age-dependent higher incidence of systemic comorbidities. Coupled with the misconception that NMOSD is a disorder exclusive of the young, atypical presentations might go undiagnosed. Intractable vomiting and hiccups are likely to be attributed to a primary gastrointestinal disorder, and even imaging suggestive of LETM may be interpreted as suggestive of a neoplasm. Our patient presented with these symptoms and only after she developed signs of a myelopathy was the diagnosis of NMOSD entertained.
LONMOSD may have a more severe phenotype. In one of the earliest series of patients with LONMOSD, the characteristics of 45 people were reviewed.Reference Camara-Lemarroy and Burton2 LONMOSD was seen predominantly in women (80%), and rapid accumulation of disability as well as a high mortality rate was noted. Another series of 30 cases of LONMOSD in a Chinese population found a median age of onset of 57.5 years (86.7% were women) and also confirmed a higher degree of disability.Reference Mao, Yin and Zhong3 Neither of these two cohorts reported APS as a presenting symptom. In the most recent series of 69 people with LONMOSD, presentation was not different from EONMOSD, but older age at onset did predict worse functional outcome (particularly in AQP4-positive patients).Reference Sepulveda, Delgado-García and Blanco4 Only six of these patients had a brainstem syndrome, but it is unclear if any these presented with APS, suggesting that brainstem involvement and APS are rare in this subpopulation.
In another recent Chinese cohort of 59 people with LONMOSD, it was noted that lesions around the 4th ventricle were less common when compared with EONMOSD.Reference Zhang, Yang and Li5 They did, however, report 12 clinical events of APS. A Korean cohort of 45 patients with LONMOSD also confirmed that brainstem lesions adjacent to the fourth ventricle and periependymal lesions surrounding the lateral ventricles were less frequently observed when compared with EONMOSD (3 vs 28 lesions).Reference Seok, Cho and Ahn6
Around 15–30% of NMOSD are considered LONMOSD at presentation, with a median age of onset between 50 and 60 years.Reference Camara-Lemarroy and Burton2–Reference Seok, Cho and Ahn6 “Very-Late NMOSD,” variably defined as with an onset >65 or 70 years of age, is rarer still. In the Chinese cohort described by Zhang et al.,Reference Zhang, Yang and Li5 only 6 out of 59 cases with LONMOSD were over the age of 65, while in the Korean cohort described by Seok et al.,Reference Seok, Cho and Ahn6 only 5 out of 45 were over the age of 65. The oldest person reported with LONMOSD had an onset at 88 years of age.Reference Krumbholz, Hofstadt-van Oy and Angstwurm7 Nevertheless, definitions of LONMOSD are inherently arbitrary as age cut-offs are not part of the diagnostic criteria of NMOSD, and the clinical utility of drawing an age distinction is unclear.
Acute management in LONMOSD does not differ from EONMOSD. Our patient responded poorly to steroids, but PLEX led to quick arrest of her worsening and after 5 cycles there was objective improvement. Use of long-term immunotherapy has been described to be less frequent in LONMOSD,Reference Sepulveda, Delgado-García and Blanco4 but authors agree that early aggressive management is warranted considering the poor outcome and rapidly accumulating disability.Reference Seok, Cho and Ahn6 Azathioprine and mycophenolate mofetil are usually employed,Reference Seok, Cho and Ahn6 but some series suggest that this subpopulation exhibits more frequent tolerability issues as well as opportunistic infectious complications.Reference Camara-Lemarroy and Burton2,Reference Krumbholz, Hofstadt-van Oy and Angstwurm7 In our patient, plans were made for long-term immunomodulation with rituximab.
Disclosures
The authors have no conflicts of interest to declare.
Statement of Authorship
AK and BB gathered the case information and performed literature review. TB and W-QL revised content of manuscript and edited the final version. CRC-L wrote the manuscript and conceptualized the report.

