



Introduction
In the past century, human life expectancy has increased exponentially in parallel with global physical inactivity and high-caloric diets across the globe (Refs Reference Partridge, Deelen and Slagboom1, Reference Burman2). According to recent projections, more than 20% of the worldwide population will exceed the age of 60 in 2050 (Ref. Reference Partridge, Deelen and Slagboom1). Concerningly, the overlap of physically inactive lifestyles and high caloric diets will increase the prevalence of obesity among older people (Ref. Reference Burman2). Thus, factors that contribute to lower health spans and poor quality of life in older age such as obesity and physical inactivity should be carefully considered. Over the last three decades some promising interventions and many preclinical studies have been found to slow the aging process and increase the lifespan of several organisms from yeast flies and rodents to nonhuman primates (Refs Reference Partridge, Deelen and Slagboom1, Reference Kaushik and Cuervo3). However, in humans, the development and prevalence of many age-related diseases are a challenge for some therapeutic interventions and thus, many knowledge gaps remain.
Poor physical fitness and low physical activity level were associated with older age and cardiovascular risk factors (Refs Reference Suominen4–Reference Guseman6). Further, reduced physical activity, low cardiorespiratory fitness, and poor muscle strength are associated with cardiometabolic disease (Ref. Reference Carnethon, Gulati and Greenland7). In contrast, regular exercise training preserves physical performance and cardiometabolic health (Ref. Reference Fridh5). In general, the cellular mechanisms implicated in prolonged lifespan tend to decrease in effectiveness with advanced age (Ref. Reference López-Otín8). Therefore, there is no surprise that aging per se is a major risk factor for many dysfunctional diseases (Ref. Reference Liu9). First described by Franceschi et al. (Refs Reference Franceschi10–Reference Franceschi, Bonafè and Valensin12), both chronic, low-grade inflammation, known as ‘inflammaging,’ and age-related changes in the immune system, known as immunosenescence, are now recognised as hallmarks of the defective aging immune system (Ref. Reference Santoro13).
Obesity has also revealed alterations in the aged immune system impacting anti-inflammatory mechanisms (Refs Reference Pirola and Ferraz14, Reference Connaughton15) and obesity-driven low-grade chronic inflammation. Obese older people's immune systems display an ‘immunosenescence’ phenotype (Refs Reference Shirakawa16, Reference Salvestrini, Sell and Lorenzini17), where immune cells become more broadly inflammatory and their cell's specific functionality is altered. In addition, to this immunometabolism age-impaired condition, a dysfunctional autophagy process has been linked to other inflammatory-based metabolic conditions (e.g., diabetes), auto-immune diseases, neurodegenerative disorders, and several cancers (Ref. Reference Deretic18). Indeed, when the autophagy process is disrupted, there is a higher risk of infections and inflammation conditions due to the accumulation of detrimental exogenous hazards, and/or endogenous cellular cytotoxic products (Ref. Reference Levine, Mizushima and Virgin19). Therefore, autophagy is a crucial process in old organisms to protect from age-related immunosenescence product accumulation (Ref. Reference Lopez-Otin20).
Regarding T cells, CD4+ T cells are key immune cells that determine the development of chronic inflammatory diseases (Ref. Reference Merkley21). CD4+ T cells orchestrate adaptive immune responses by producing cytokines and effector molecules. These functional roles of T cells vary depending on the surrounding inflammatory or anatomical environment (Ref. Reference Jeong, Choi and Lee22). Autophagy is an important process that regulates the function of CD4+ T cells by clearing dysfunctional cytoplasmic materials for lysosomal degradation, and autophagy CD4+ T cell-mediated immune responses, including cytokine production, proliferation, and differentiation. Furthermore, through canonical processes involving autophagy machinery, autophagy may also contribute to the development of chronic inflammatory diseases. Therefore, a targeted therapeutic intervention of autophagy processes could be used to treat several chronic inflammatory diseases (Refs Reference Jeong, Choi and Lee22, Reference Watanabe23). In this perspective review, we will first provide an overview of the autophagy process and then, we will discuss the impact of adipose tissue accumulation and physical fitness in autophagy in naïve CD4 T cells in aging.
Autophagy process and types – overall view
Autophagy is the ‘clearance’ cellular process that destroys dysfunctional and/ or unnecessary intracellular components for quality control, attenuates stress effects and maintain cellular homoeostasis. This catabolic process is lysosome-dependent whereby damaged proteins or long-lived organelles are broken down and then recycled to create adenosine triphosphate (ATP) and/or novel organelles (Ref. Reference Levine, Packer and Codogno24). Autophagy is upregulated in different physiological stress conditions or stimuli including in hypoxia states, oxidative stress (OS), amino acid starvation, energy deprivation, DNA damage, intracellular pathogens and protein aggregation (Ref. Reference Kovacs25). During these physiological conditions, autophagy recycles cytoplasmic substances and upregulates cell survival mechanisms (Ref. Reference Merkley21). Therefore, overall autophagy is considered an endogenous protecting mechanism.
Most cells maintain slight basal autophagy activity to survive in normal conditions but cells have also an apoptotic function called autophagic cell death, depending on specific conditions (Ref. Reference Fitzwalter and Thorburn26). Autophagy is activated and regulated by a variety of cellular components including the autophagy- related proteins (ATG) family members and the mammalian target of rapamycin (mTOR) kinase signalling pathways. Overall, the most important complexes modulating autophagy are mTORC1, the Unc-51 like the autophagy activating kinase 1 (ULK1 also known as ATG1), the autophagy-related protein 13 (ATG13) complex, complexes containing phosphoinositide 3 kinase (PI3 K) and its related proteins like VPS34 that is encoded by PIK3C3 (Refs Reference Liu and Levine27, Reference Madrigal-Matute and Cuervo28). ULK1 is regulated by the nutrient- and energy-sensing kinases mTORC1 and 5′ AMP-activated protein kinase (AMPK). In addition to the classic mTOR–ULK1 complex and PI3 K signalling pathways, other cell stress signals, such as extracellular- signal regulated kinase (ERK), JUN N- terminal kinase (JNK) and p53 also contribute to the modulation of autophagy activity in reaction to physiological and environmental stressors (Ref. Reference Cheng29). The autophagy signalling starts with activation of ULK1 complex that phosphorylates Beclin-1 and PI3 K complex (Ref. Reference Russell30). This process triggers the recruitment of ATG to form the autophagosome. Three different types of autophagy have been recognised, including macro-autophagy, micro-autophagy and chaperone-mediated autophagy (CMA) (Ref. Reference Macian31).
Macro-autophagy relies on de novo formation of autophagosomes, to sequester and transport cargo to the lysosome and can be either selective or nonselective (Ref. Reference Parzych and Klionsky32). Selectivity is mediated by autophagic receptors connecting cargo to the growing phagophore. Autophagy receptors link cargo to phagophores by attaching to ATG8 genes via light chain 3 (LC3)-interacting region (LIR) motifs. For instance, the neighbour of BRCA1 gene 1 (NBR1), the sequestosome 1–like receptors (SQSTM1)/ p62, TAX1BP1, OPTN and NDP52 bind ubiquitylated cargo such as damaged organelles and protein aggregates, as well as intracellular bacterial pathogens (Ref. Reference Kimura, Mandell and Deretic33). There are also specific organelle-bound receptors to operate autophagy in mitochondria and endoplasmic reticulum. ATG8s are covalently linked to the E3-ubiquitin ligase–like complex ATG16–ATG5–ATG12 and are thus capable of binding to the membrane of the phagophore. In mammalian cells, most ATG genes are seen on isolation membranes (e.g., ATG13, ULK1/2, FIP200, Beclin 1, ATG101, ATG14, ATG12 and ATG16L1) but not on thorough autophagosomes (Ref. Reference Longatti and Tooze34). Mammals have seven ATG8 isoforms; GABARAP, GABARAPL1, GABARAPL2, LC3A, C, -B and -B2 (Ref. Reference Mejlvang35). However, to date, only microtubule-related protein LC3, a mammalian homologue of ATG8 in yeast, is known to be on autophagosomes, and thus, this gene serves as an indicator for autophagosomes and LC3 B is a good index for autophagic activity (Ref. Reference Mizushima36).
Micro-autophagy involves the direct uptake of cargo through the invagination of the lysosomal membrane, forming vesicles within the lysosome and the autophagic cargoes which are then degraded in the endolysosomal lumen (Refs Reference Parzych and Klionsky32, Reference Wang, Klionsky and Shen37). Like macro-autophagy, micro-autophagy can be selective and non-selective or cargo-specific depending on the cellular milieu and is the main autophagic response under starvation and refeeding conditions but can also be induced by nitrogen starvation or rapamycin via regulatory signaling complex pathways (Ref. Reference Li, Li and Bao38). Currently, there are two different types of micro-autophagy processes proposed based on autophagy machinery: (1) fission-type micro-autophagy, which is independent of the core autophagy machinery and is mediated by endosomal sorting complexes required for transport that mediate membrane scission or budding (Ref. Reference Wang, Klionsky and Shen37); and (2) fusion-type micro-autophagy that requires the core autophagy machinery and soluble N-ethylmaleimide-sensitive factor attachment protein receptor complexes (Ref. Reference Schuck, Gallagher and Walter39). The most studied type of micro-autophagy is the fission type but both micro-autophagy mechanisms are not fully determined.
Chaperone-mediated autophagy is a lysosomal pathway of proteolysis that is responsible for the degradation of cytosolic proteins under prolonged nutrient deprivation (Ref. Reference Dice40) and or oxidative stress but it is a process that can be also inhibited by glucose-6-phosphate dehydrogenase and the heat shock protein of 90 (HSP90). This autophagy process involves the straight uptake of cargo by lysosomes dependent on lysosomal membrane protein 2A as well as chaperone HSC70 (Ref. Reference Cuervo and Wong41). In contrast with macro-autophagy and micro-autophagy, chaperone-mediated autophagy does not require vesicular traffic (Ref. Reference Majeski and Dice42) but in turn, requires cytosolic proteins with particular peptide sequence motifs to be recognized by molecular chaperones and delivered to lysosomes as extensively reviewed by Parzych and Klionsky (Ref. Reference Parzych and Klionsky32). Despite quite distinct, all three types of autophagy have the same final purpose- delivery of cargo to the lysosome for degradation and recycling, allowing a positive cellular renewal. In turn, dysfunctional autophagy is associated with various human diseases (Ref. Reference Parzych and Klionsky32). Thus, finding therapeutic strategies that enhance endogenous autophagy are now a promising therapeutic avenue to manage/revert unbalanced functions such as apoptosis, inflammation and other intracellular processes in several inflammatory conditions including ischaemic heart disease, rheumatic conditions, neurodegeneration and tumours (Refs Reference Wang43, Reference Rockel and Kapoor44) Figure 1.

Fig. 1. Illustrative age-related type and process of autophagy. Created with BioRender.com.
Autophagy in aging – targeting naïve CD4+ T cells
Aging is a natural phenomenon characterised by the progressive and gradual turnover of cellular components including genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulation of nutrition-sensing, mitochondrial dysfunction, cellular senescence, cellular exhaustion and altered intercellular communication- also known as the ‘hallmarks of aging’ (Refs Reference López-Otín8, Reference Hernandez-Segura, Nehme and Demaria45). Interestingly, malfunction or disruption of autophagy in old organisms plays a crucial role in these cellular age-related dysfunctions (Ref. Reference López-Otín8). Multiple autophagy signalling pathways and the lysosomal compartment are affected by aging, and together contribute to the inefficient induction and autophagy degradation (Ref. Reference Kaushik46). Autophagy also contributes to the regulation of other basic mechanisms of aging, including modulation of cellular metabolism and nutrient sensing pathways, maintenance of stemness properties in cells, regulation of epigenetic signatures, repair of macromolecular damage, prevention of organelle dysfunction and inflammation and cellular response to different forms of stress (Refs Reference López-Otín8, Reference Lopez-Otin20, Reference Hernandez-Segura, Nehme and Demaria45).
In terms of the immune system, autophagy operates as a ‘stimulator’ that influences both the adaptive and innate immune systems and regulates antigen presentation, thymic selection, cytokine production and development of T cells survival and homoeostasis (Ref. Reference Kuballa47). Autophagy also modifies T cells functions by mediating the differentiation and activation of effector and memory CD4+ T cells phenotype, as well as its metabolic demand which collectively, modulates immunological responses (Ref. Reference Harris48). Thus, autophagy is a mechanism of intracellular pathogen sensing, and deficits in autophagy can cause enhanced susceptibility to infection. Autophagy also modulates cell-specific pattern-recognition receptors signalling and inflammasome, as well as the clearance of apoptotic corpses, potentially as a process to regulate inflammation (Ref. Reference Jia and He49). Therefore, autophagy distinctly modulates different immune system cellular components and functions.
Recent findings suggest that autophagy-deficient immune cells display features of premature aging, while aged T cells have decreased autophagy (Ref. Reference Linton and Thoman50). Advanced age also leads to poor T cell function, to suboptimal antibody and cytokine production and to increases in cytotoxic T cells in response to immunisation (Refs Reference Linton and Thoman50, Reference Goronzy and Weyand51). Changes in the composition and loss of function in CD4+ T cells populations are also well described with aging, although the underlying mechanisms remain unclear (Ref. Reference Chang, Jensen and Hurley52). Adaptive immunosenescence is characterised by loss of receptors, decreased repertoire diversity, impaired effector responses against new antigens and impaired immunological memory function (Ref. Reference Linton and Thoman50). Poor proliferation and diminished responsiveness to IL-2 appears to be a ubiquitous feature of aged T cells, and may explain the delayed peak in T cell expansion and cytotoxic activity commonly observed in the elderly.
Evidence suggests that changes in autophagy with aging are not mostly due to decreased ATG gene expression but rather to age-related effects on immune system composition and/or to the defects in signalling (Ref. Reference Goronzy53). In the thymus, naïve CD4 T cells are quiescent, and for instance, in young organisms, the majority of cells in the CD4+ T cells pool have a naïve cell surface phenotype (CD44lowCD62Lhigh) and stringent antigen and co-stimulatory requirements, as well as a delay before the onset of proliferation after antigen recognition (Ref. Reference Cambier54). In contrast, only a smaller cohort of these younger CD4+ T cells pool results from previous exposure to antigen and has a different reciprocal memory phenotype (CD44highCD62Llow). These memory cells respond more rapidly to physiological stress and have less stringent requirements for antigen and co-stimulatory factors than naïve CD4+ T cells. During physiological stress, naive CD4+ T cells are activated after interaction with antigen-MHC complex and differentiate into specific T cell subtypes depending mainly on the cytokine milieu of the microenvironment. Advancing age alters this youth phenotype, leading to a predominance of CD44highCD62Llow cells that are potentially the accumulative effect of a lifetime of exposure to antigens (Refs Reference Clise-Dwyer55, Reference Dobber56). Therefore, the proportion of naïve T CD4+ cells decreases (Ref. Reference Haynes, Linton and Swain57) and the production of naïve CD4+ T cells effectors reduces, as result, at least in part, of the defective production of IL-2 (Ref. Reference Haynes58)- an age-related effect likely due to the aging of individual T cells and its cytokine production (Refs Reference Clise-Dwyer55, Reference Hale59). Thus, to overcome this cellular impairment, an additional mechanism that contributes to restore naïve T CD4+ cells numbers has been termed homoeostatic proliferation (Ref. Reference Fülöp60). Still, the molecular mechanisms that establish and maintain naïve T CD4+ cells numbers remain elusive.
Aged naïve T CD4+ cells are suggested to be anergic (unresponsive), and this defective response can lead to either reduced clonal burst size or increased apoptotic death (Ref. Reference Eisenbraun, Tamir and Miller61). Previous studies have reported that activated aged T cells are less susceptible to Fas–FasL mediated death (Refs Reference Gupta and Gollapudi62–Reference Sharma64). Fas ligand (CD95L) is a type-II membrane protein within the tumour necrosis factor (TNF) superfamily of death receptors (Ref. Reference Suda65). FasL shares sequencies homology with tumour necrosis factor-alpha (TNF-α) and TNF-related apoptosis-inducing ligand, with similarity present in the C-terminal homology ectodomain that extends into the extracellular space for binding the receptor (Ref. Reference Locksley, Killeen and Lenardo66). FasL engages and trimerises the death receptor Fas (CD95) on cell surfaces to initiate the extrinsic apoptosis pathway (Refs Reference Paulsen and Janssen67, Reference Strasser, Jost and Nagata68). Thus, the Fas-FasL interaction recruits the Fas-associated death domain adapter protein (FADD) via death domain binding and procaspase-8 to form the death-inducing signalling complex (Refs Reference Magnusson and Vaux69, Reference Marsters70).
Activating the caspase-8 pathway catalyses its autoactivation, and subsequently the proteolytic conversion of downstream effector caspases such as caspase-3 and −7 into their mature forms (Ref. Reference Boatright and Salvesen71). Effector caspases direct cell death by apoptosis, which results in nuclear and cytoplasmic condensation followed by cellular fragmentation into membrane-bound apoptotic bodies (Ref. Reference Perl72). On the other hand, in aged humans, naïve CD4+ T cells, central memory and effector memory cells are reported to be more susceptible to both Fas-mediated and TNF-α-mediated apoptosis (Refs Reference Gupta and Gollapudi62, Reference Gupta and Gollapudi73). Taken together, naïve CD4+ T cell regulation can be boosted by the induction of autophagy, which also helps to control systemic chronic inflammation in the elderly. Furthermore, the effector functions of these cells are mediated by the cytokines secreted by the differentiated cells. Therefore, apart from the positive endogenous regulatory quality control mechanism, autophagy can be seen as a stimulator of naïve CD4+ T cells and its downstream effector cells in advanced age- an anti-inflammaging process (Ref. Reference Conte74). Compared to age-matched controls, naïve CD4+ T cells from old individuals with extended longevity and healthy aging show preserved autophagic activity (Ref. Reference Gupta and Gollapudi73). In contrast, defects in autophagy shortage are associated with disruption on different functional processes of naïve CD4+ T cells like differentiation, maturation, cell death which are associated with an increased risk of age-related immune or inflammatory complications (Ref. Reference Mattoo75) Figure. 2.

Fig. 2. Schematic age-related autophagy pathway in naïve CD4+ T cells. Age-related possible intrinsic and extrinsic mechanisms. Created with BioRender.com.
Potential therapeutic modulators targeting Naïve CD4 T cells
The immunological function of autophagy represents the interaction of this cellular process within a single cell, such as pathogen elimination in antigen-presenting cells (APC), and proliferation through clearance of cell cycle proteins in naïve CD4+ T cells (Ref. Reference Mattoo75). However, several promising candidate drugs and modulators can potentially activate or inhibit autophagy processes targeting naïve CD4+ T cells, specifically relying on the nucleation, elongation, fusion, or degradation phase.
Metformin (Refs Reference Bharath76–Reference Saisho78), a well-tolerated type 2 diabetes (T2D) drug that improves glycemic control and, in some studies, chronic inflammation (Refs Reference Malínská79, Reference Cameron80), is an autophagy activator candidate. Bharath et al. (Ref. Reference Bharath76) reported a combinatorial cytokine profile differentiated CD4+ T cell inflammation in healthy sexagenarians compared to 30-year-old subjects. Safe and physiologically achievable concentrations of metformin decreased overall T cell-induced cytokine production ex vivo in samples from all subjects, however, the differentiated Th17 cytokine profile was more prominent in older individuals and decreased the differentiated Th2 cytokine profile in younger individuals. Furthermore, metformin increased autophagy in CD4+ T cells in older individuals and shifted mitochondrial bioenergetics and T cell inflammation, but not mitophagy, in young individuals' cells. In older individual cells, mitochondrial function was compromised and activated a Th17 profile from T cell profiles was produced.
Lipids control the activity of several key signaling molecules of naïve CD4+ T cells activation (Refs Reference Lochner, Berod and Sparwasser81, Reference Weitz-Schmidt82). Statins are a family of chemically related small molecules, which inhibit hydroxy-methyl-glutaryl Coenzyme A (HMG-CoA) reductase, a rate-limiting enzyme that catalyzes the conversion of HMG-CoA to mevalonate, a key intermediate in the cholesterol biosynthetic pathway (Ref. Reference Ghittoni83). Weitz-Schmidt et al. (Ref. Reference Weitz-Schmidt82), observed that a different subset of statins, such as lovastatin and simvastatin, but not mevastatin and pravastatin, inhibit naïve CD4+ T cell activation. Interestingly, the authors reported that this activity was found to be independent of the capacity of these statins to inhibit HMG-CoA reductase, but resulted from statin binding to lymphocyte function-associated antigen-1 (LFA-1), a heterodimeric glycoprotein belonging to the b-2 integrin family and constitutively expressed in an inactivate state on the surface of CD4+ T cells in adults hypercholesteremic individuals (Refs Reference Weitz-Schmidt82, Reference van Seventer84).
Data from several models of organisms suggests that mTOR is a critical controller of aging (Refs Reference Evans85, Reference Robida-Stubbs86), and rapamycin treatments can extend the lifespan of mice (Ref. Reference Harrison87). The mTOR signaling pathway is downstream of many growth factor receptors and regulates cell proliferation, growth, differentiation, and survival (Ref. Reference Galluzzi88). It continuously inhibits autophagy, and compounds that interfere with kinases within this pathway are potent inducers of autophagy (Ref. Reference Kim and Guan89). In this sense, Perkey et al. (Ref. Reference Perkey90) hypothesized if long-term rapamycin treatment to mTOR inhibition would prevent some of the age-related changes in CD4+ T cells. The authors observed that aging decreases the number of CD4+ T cells expressing high levels of pro-apoptotic markers, such as BIM and pERM when compared with CD4+ T cells from young mice. Moreover, untreated CD4+ T cells from old mice showed more cells with higher levels of phosphorylated AKT. Rapamycin resulted in diminished age-related increases in the proportion of memory cells as indicated by the CD44 and CD62L markers as well as demonstrating that rapamycin affects CD4+ T cell differentiation (Refs Reference Perkey90, Reference Powell91).
Nutritional modulators have been extensively related to autophagy targeting CD4+ T cells. Caloric restriction, for example, represents a substantial reduction in caloric intake about 30 to 60 % below the ad libitum levels, while maintaining adequate nutrition, improving health, and extending lifespan in many organisms, including primates, through a mechanism that is not understood (Refs Reference Colman92–Reference Kemnitz94). The lack of mTOR homolog TOR1 has an extended replicative lifespan that is not further enhanced by caloric restriction, which suggests a common mechanism (Ref. Reference Liang95). Farazi et al. (Ref. Reference Farazi96) reported the ability of agonist OX40 treatment to be effective in improving tumor-free survival in young tumor-bearing mice, but the same effect was not observed in older 12-month or 20-month-old tumor-bearing mice. Wang et al. (Ref. Reference Wang97) reported that caloric restriction resulted in higher production of IL-2 in CD4+ T cells, as indicated by the presence of more IL-2-producing CD4+ T cells.
Overall, these autophagy modulators might lead to important therapeutic avenues to optimize the effect of immune-based interventions in older individuals, particularly modulators targeting naïve CD4+ T cells.
Autophagy and adipose tissue
Obesity has increased markedly over the past decades and reached epidemic proportions. By 2050, over one-third of adults will be overweight (BMI 25–29.9 kg/m2) or obese (BMI ≥ 30 kg/m2) worldwide (Refs Reference Johnson98, Reference Bastien99), a reflection of the modern lifestyle of high-caloric food intake and reduced physical activity. The hallmark of obesity is the accumulation of dysfunctional adipose tissue when energy intake exceeds energy expenditure, which triggers metabolic stress. This metabolic process increases inflammatory responses and levels of fatty acids, triglycerides and low-density lipoprotein cholesterol (LDL-c) resulting in a cluster of interconnected conditions that predispose to metabolic syndrome, cardiovascular diseases, musculoskeletal pathologies, cancer and neurodegenerative diseases (Refs Reference Afshin100, Reference Hubler and Kennedy101). In addition, chronic exposure to food overload can lead to immunometabolic deterioration of adipose tissue. Resident adipose tissue macrophages, which in homoeostasis are maintained in an M2 polarisation state by local interleukin-4, 13, and 10 (IL-4, IL-13 and IL-10), become activated and lose their regulatory and anti-inflammatory properties (Refs Reference Ross, Devitt and Johnson102, Reference Lin and Wei103).
In obesity, there is an imbalance between increased food intake and inadequate energy expenditure, leading to suppression of autophagy due to increased mTOR signaling- a major repressor of autophagic activity (Ref. Reference Zhang, Sowers and Ren104). In contrast, caloric restriction promotes optimal health and delays pathological processes upregulated by adaptive immune system stress signaling cascades, improving mitochondrial health, DNA repair, and autophagy (Refs Reference Katsi105–Reference Mattson, Longo and Harvie107). It has also been reported that caloric restriction might activate hypoxia-inducible factor 1 (HIF1), which is known to facilitate transactivation of BCL-2/adenovirus E1B protein-interacting protein 3 (BNIP3) and BNIP3-like (BNIP3L) to stimulate mitophagy (Ref. Reference Bellot108). Autophagy has also been shown to be altered in adipose tissue in obese individuals (Refs Reference Soussi, Clément and Dugail109, Reference Haim110).
Basal autophagy is essential for the maintenance of cellular and organismal homoeostasis (Ref. Reference Juárez-Rojas111). Thus, aberrant changes (increases or decreases) in autophagy contribute to the pathogenesis of various human diseases. Interestingly, alterations in autophagy process (enhanced or suppressed) have been reported in both: patients with obesity and animal models of genetically predisposed or diet- induced obesity (Refs Reference Ignacio-Souza112, Reference Jaishy113). Global and tissue- specific deletion and/or mutation of certain autophagy genes or autophagy regulatory molecules result in altered lipid metabolism, hepatic steatosis and an obese or diabetic phenotype in animal models (Ref. Reference Lee114). It has been well established that autophagy selectivity depends on the type of stress; for example, mitochondrial stress promotes autophagy of damaged mitochondria (mitophagy), whereas lipid overload facilitates autophagy of lipid droplets (lipophagy) (Ref. Reference Kim and Lee115).
Autophagy is sensitive to changes in nutrient status, particularly high-fat (HFD) and/or excess caloric intake. More importantly, changes in autophagy might unfavourably influence metabolism locally (in a metabolically active organ) or globally to promote metabolic dysregulation through endocrine mechanisms (Ref. Reference Zhang, Sowers and Ren104). For instance, dyslipidemia, high blood pressure and increased blood levels of glucose, which represent three major risk factors in obesity, are believed to be responsible for unfavourable pathological outcomes and changes in autophagy during obesity (Ref. Reference Oga and Eseyin116). Rodents with whole- body or tissue- specific (liver and pancreas) deletion of the BECN2, ATG7, LAMP2, TFEB, and BIF genes have obesogenic metabolic phenotypes or are predisposed to HFD- induced or genetic (leptin- deficient (ob/ob)) obesity (Ref. Reference He117). In addition, liver- specific knockout autophagy genes ATG7 and TFEB promote liver steatosis and weight gain, whereas adenoviral overexpression of ATG7 and TFEB protected against weight gain and metabolic syndrome (Ref. Reference Liu118).
Regarding the effect of obesity on immune system, obesity-associated changes affect both innate and adaptive immunity, dramatically impairing immunological responses against infection (Ref. Reference Jain and Chaves119). Adipocytes and macrophages are thought to be primary sources of proinflammatory mediators in obesity and are important in disease pathogenesis (Ref. Reference Duffaut120). Emerging evidence has suggested the involvement of distinct lymphocyte subpopulations during obesity-associated inflammation, reflected by higher memory and lower naïve CD4+ T cells proportions, which is associated with prevalent type 2 diabetes in a multi-ethnic cohort study (Ref. Reference Olson121). Findings associating higher memory and lower naïve CD4+ T cells with type 2 diabetes are consistent with the role of chronic adaptive immune system activation and exhaustion seen during increased inflammation in type 2 diabetes (Refs Reference Olson121–Reference McLaughlin123). This relationship between higher memory and lower naïve CD4+ T cells with type 2 diabetes may reflect both direct and/or indirect mechanisms by diabetogenic or obesity-associated antigens (Ref. Reference Winer122).
Lipids (e.g., fatty acids and cholesterol) are potent modulators of T cell function and in pre-clinical and cell culture studies findings suggest that high lipid load may directly contribute to obesity-associated immune dysfunction (Ref. Reference Gorjão124). Several studies support that autophagy is regulated by intra- and extracellular lipid levels (Refs Reference Kim and Lee115, Reference Niso-Santano125, Reference Koga, Kaushik and Cuervo126) and that autophagy is inhibited by high lipid concentrations (Refs Reference Koga, Kaushik and Cuervo126, Reference Mei127). The specific mechanisms of how lipids influence autophagy are still not completely understood. In this context, Guerrero-Ros et al. (Ref Reference Guerrero-Ros128) have reported that high lipid levels negatively affect the activation-induced autophagy in T helper cells, which contributes to the inhibition of T cell responses observed in CD4+ T cells exposed to increasing concentrations of fatty acids or isolated from diet-induced obese mice (Ref. Reference Guerrero-Ros128). This study also showed a dose-dependent reduction in the rate of accumulation of LC3-II in cells exposed to oleic acid, supporting that lipid challenge may cause an inhibition of autophagosome formation in T cells (Ref. Reference Guerrero-Ros128). Similarly, in a preclinical study with a CD4+T cell specific KLF10 knockout model, a member of the Kruppel-like transcription factor family, Wara et al. found an association with obesity, insulin resistance and fatty liver (Ref. Reference Wara129). These obesity associated changes were due to impairments of CD4+ Treg cells mobilisation to the liver and adipose tissue (Refs Reference Kalathookunnel Antony, Lian and Wu130, Reference Misumi131). Physiologically, these knockout cells had reduced mitochondrial respiration and glycolysis, and reduced PI3 K-Akt-mTOR signalling activation, which is simultaneously an important autophagy activation complex (Ref. Reference Misumi131). Unfortunately, more research is needed to determine the underlying mechanisms that disrupt autophagy in CD4+ T cell homoeostasis, activation and metabolism once evidence in obese individuals is very limited.
Aged but fit – targeting autophagy
Environmental conditions such as caloric restriction and physical exercise modulate autophagy, a process that is ‘turned on’ under these stress conditions, recycling long-lived or damaged cellular organelles and proteins for the resynthesis of ATP and new organelles (Ref. Reference Ren, Sowers and Zhang132). Changes in physical exercise status, alter autophagy sensitivity to a more effective metabolism locally or globally (Ref. Reference Zhang, Sowers and Ren104). Physical exercise stimulates the expression of crucial genes, such as AMPK and Parkin, and inhibits mTOR signalling cascade, increasing cell autophagic activity. Mechanistically, autophagy follows canonical pathways in which the activation of AMPK inhibits mTOR signalling pathway (Ref. Reference He133). Historically, the first report of a potential function of macroautophagy in exercise- related adaptive response was acknowledged in research that reported a shifting of LC3-I to LC3-II and a decline in p62 content in skeletal muscle after a single bout of aerobic training (Ref. Reference He133). Similarly, this switch of LC3-I to LC3-II, coupled with the decline in the expression of p62 and increment in dynamin related protein 1(DRP1) expression (i.e., a mitochondrial fission protein) were also seen in a rodent myocardium following a single bout of aerobic exercise (Refs Reference He133, Reference Li134). These exercise related changes impact autophagy once the cytosolic LC3-I is converted to LC3-II form after being cleaved by ATG-4, which is activated by ROS during physical exercise (Ref. Reference Wu135). This conversion is needed to recruit autophagosomal membranes and leads to the formation of autophagosomes (Ref. Reference Li134). Thus, (in)directly exercise enhances mitophagy—a selective autophagy process which in turn, allows cellular clearance of dysfunctional mitochondria and reduces ROS production leading to a favourable local and systemic global milieu (Refs Reference Li134, Reference Wu, Zhang and Ren136, Reference Biala, Dhingra and Kirshenbaum137).
Autophagy has been shown to prevent senescence partly due to its ability to maintain mitochondrial fission and fusion homoeostasis and prevent ROS (Ref. Reference Korolchuk138). Acute exercise increases mitophagy depending on the phosphorylation of AMPK and ULK1 complexes. In turn, AMPK signalling pathway is activated by exercise-associated incline on AMP/ATP ratio and by sympathetic activation (Ref. Reference Wu135). AMPK also affects mitochondrial biogenesis by modulating PGC-1a expression. In normal conditions, mitophagy starts when injured mitochondria are marked for degradation. The main fission protein Drp1 replaces the depolarised mitochondrial membrane and separates the damaged portions, from the rest of the ‘normal’ mitochondria. Then, PTEN induced kinase 1 (PINK1) recruits E3 ubiquitin-protein ligase Parkin that ubiquitinates proteins branch on the external mitochondrial membrane (Ref. Reference Saito and Sadoshima139). Thereafter, PARKIN gene increases transcription to target mitochondria to autophagosome by attaching LC3 on phagophore in response to promotor signal (autophagy stimulator) (Ref. Reference Antón140). Still, despite PARKIN indispensable role in initiating exercise-induced mitophagy, in a Parkin knockout model, exercise did not affect basal mitophagy (Refs Reference Chen, Erlich and Hood141, Reference Fix142), suggesting that exercise-induced mitophagy triggers other signalling pathways. Interestingly, PINK1 can activate receptors of autophagy with a light rate, independently of PARKIN gene activation (Ref. Reference Lazarou143).
Clinical investigations have reported that high-intensity interval training (HIIT) - consisting of alternating low (~ 40% VO2max) and high intensity exercise (~80% V̇O2max) profoundly suppressed ROS and inflammation which consequently improved hemodynamic dysfunctions (Refs Reference Wang144, Reference Fu145). Moreover, Wang et al. (Ref. Reference Wang, Chen and Weng146) demonstrated that exercise training under hypoxic conditions reduced senescent T cells subsets increasing circulating IFN-γ level, which also led to decreased ROS and pro-inflammatory cytokine production. A potential mechanism involved in T cells senescence is trigged by the killer cell lectin-like receptor G1 (KLRG1) - identified as a marker of replicative senescence on human T cells (Ref. Reference Beyersdorf147) frequently coupled with a reduced expression of co-stimulatory molecule CD28 (Ref. Reference Weng, Akbar and Goronzy148). Ligation of CD28 with its cognate receptor on antigen-presenting cell (APC) upregulates the secretion of interferon-c (IFN-c) and IL-2, leading to activation and proliferation of naïve CD4+ T cells (Ref. Reference Weng, Akbar and Goronzy148).
Exercise training reduces the extent of decline in hypoxic- induced high intensity exercise in autophagy and potentiated apoptosis of CD4+ T cells, which are accompanied by decreases in ROS and circulatory T helper 2 cells subsets cytokine production (Ref. Reference Weng149). Weng et al. (Ref. Reference Weng149), reported that five weeks of HIIT and moderate continuous training improved the effect of autophagy promoted by hypoxic-induced HIIT exercise and decreased apoptosis. These hypoxic exercise-induced changes were due to decreased mTOR and phosphorylated BcL-2 levels in CD4+ T cells. Downregulated mTOR triggered autophagy to protect cells from ROS and toxicity (Refs Reference Niso-Santano149, Reference Koga, Kaushik and Cuervo150), whereas inhibiting Bcl-2 phosphorylation led to the inactivation of caspase-9 cascade and increased intrinsic apoptosis (Ref. Reference Hsieh, Athar and Chaudry151). Still, elucidating the relationship between exercise training modes and CD4+ T cells autophagy/apoptosis is needed to develop a suitable training regimen to minimise the risk of immune cell death evoked by hypoxic stress (Ref. Reference Weng149).
Interestingly, compared with a control group, McCormick et al. showed that prediabetes blunts exercise benefits on autophagy after an acute bout of aerobic exercise suggesting that greater exercise benefits may be dependent on metabolic disease, exercise training regimen duration or autophagy signalling activation (Ref. Reference McCormick152). Regular exercise largely operates via the activation of different signaling pathways including the IGF1-PI3K-Akt pathway, which contributes to cell growth, metabolic homeostasis, cellular survival, mitochondrial maintenance, and enhanced translation and transcriptional processes (Ref. Reference Wu135). Exercise also activates AMPK which has been described as a potent autophagy-inductor and it is an important glucose/ insulin homeostasis signaling pathway. AMPK regulates multiple metabolic processes and is dysregulated in major chronic diseases, such as obesity, inflammation, and diabetes (Refs Reference Jeon153, Reference Rosa-Neto154). Diabetes disrupts insulin homoeostasis which inhibits AMPK by inducing its direct phosphorylation by AKT, which in turn also disrupts the activation of autophagic signalling cascade's activation (Ref. Reference Day, Ford and Steinberg155). Notably, higher exercise intensities were more efficient to enhance AMPK activity and increase autophagy-related gene expression and autophagic flux (Ref. Reference Schwalm156). Similarly, 8-weeks of resistance exercise increased protein expression of beclin-1, ATG12, ATG16 and LAMP-2 which are major autophagy inductors and activators of autophagosome activity (Ref. Reference Mejías-Peña157). Taken together, this evidence suggests that metabolic diseases may suppress exercise autophagic benefits particularly on programmes of short-term duration. Therefore, more research is needed to fully understand the role of exercise in autophagy process on CD4+ T immune cells and effectors and untangle the modulating role of different exercise training modes (aerobic vs resistance vs concurrent), intensities (low vs moderate vs high) and duration (acute vs short-term vs long-term) in immunometabolic impaired diseases.
Immunosenescence leads to thymic atrophy and increased frequency of senescent T cells mainly by shifting the naïve/memory T cell ratio (Refs Reference Duggal158, Reference Turner159), including low numbers and proportions of naïve cells, higher numbers and proportions of late-stage differentiated effector memory cells and a shift in the CD4:CD8 ratio. Notably, both acute exercise and particularly, long-term exercise seem to ameliorate features of age-related immunosenescence (Ref. Reference Turner159). In a systematic review with meta-analysis summarizing the impact of exercise on markers of cellular immunosenescence in either young or old humans (Ref. Reference Mathot160), Dihn et al. found that even an acute bout of exercise increases the numbers of senescent, naïve, memory CD4+ and CD8+ T lymphocytes in peripheral blood (Ref. Reference Cao Dinh161). Similarly, three weeks of either concentric or excentric exercise reversed the hallmarks of T cell senescence in pre-diabetic subjects through the production and mobilization of naïve T cells (Ref. Reference Philippe162). Further, 6-weeks of low-dose combined resistance and endurance exercise training program increased the CD4+/CD8+ T cell ratio and decreased plasma levels of IL-6, IL-8, IL-10, and vascular endothelial growth factor indicating that even short-term and low-threshold exercise training can stimulate immunity. This beneficial anti-inflammatory exercise effect suggests that exercise transiently shifts the features of immunosenescence. However, the evidence is scarce in elderly people and thus, understanding how exercise modulates autophagy in CD4+ T immune cells will open a new prospect to treat immunosenescence and will help to better prescribe exercise training programs in each pathology in older age (Figure 3).
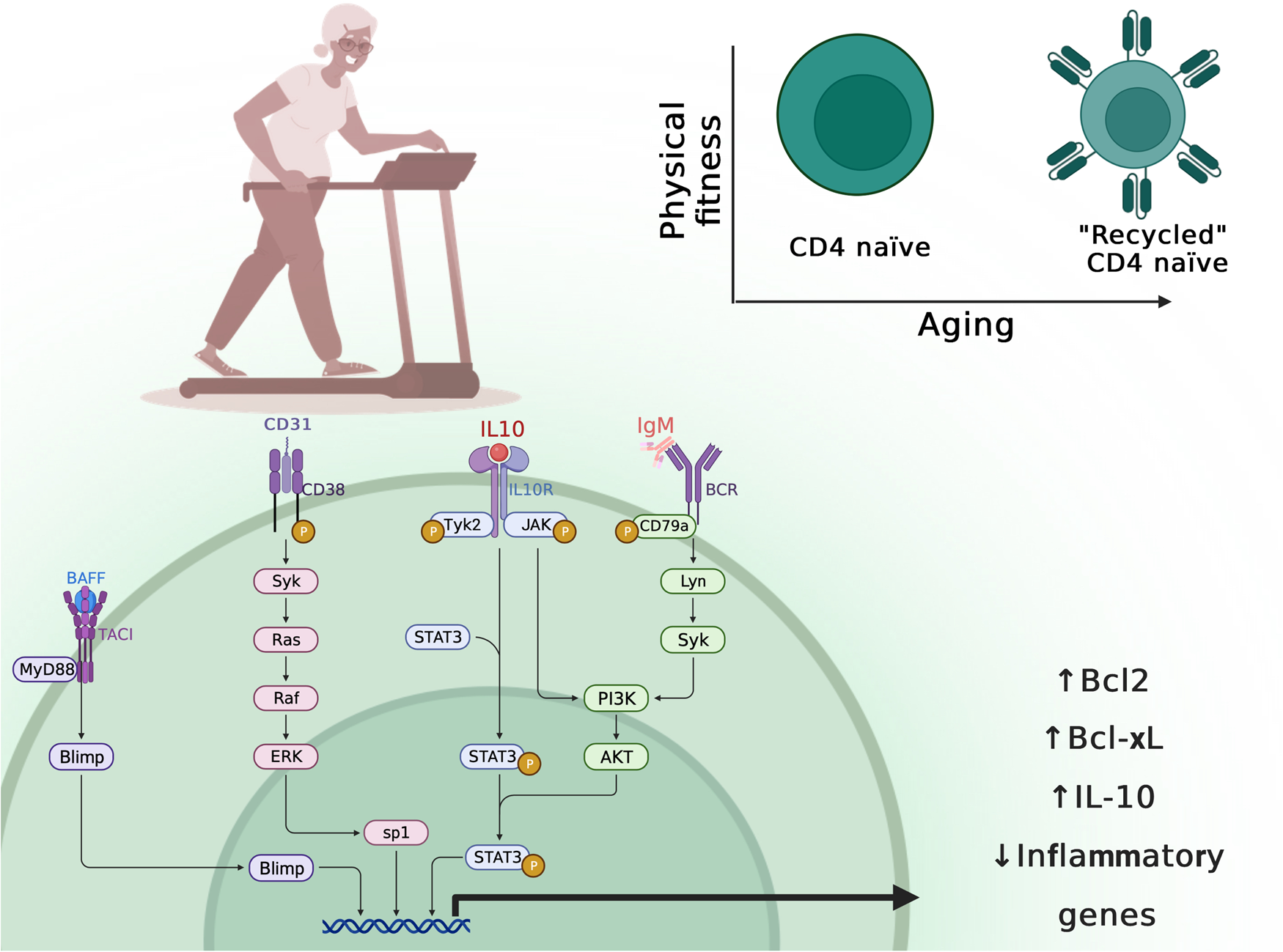
Fig. 3. Impact of physical activity on age-related ‘recycling’ naïve CD4+ T cells. Created with BioRender.com.
Translation and clinical implications
The crucial roles of different types of autophagy in various tissues have been widely studied in the last decade (Ref. Reference Lee163). Autophagy acts as an immune effector that leads to pathogen clearance. In addition, autophagy connects and regulates both the innate and adaptive immune systems and (in)directly regulates naïve CD4 T cell function, homoeostasis, activation and differentiation. Naïve CD4+T cells are essential to an effective immune response to pathogens and present as a promising route for treatment via an enhanced autophagic response. Naive CD4+T cells are activated after interaction with antigen-MHC complex and differentiate into specific subtypes depending mainly on the cytokine microenvironment. Notably, endogenous and environmental factors such as aging and obesity negatively suppress autophagy and CD4+T cells function and effector cells whereas different exercise regimens (HIIT, aerobic and resistance) have been shown to significantly improve autophagy and age-related metabolic dysfunctions. This exercise ability to improve autophagy and recycle damaged organelles (e.g., mitochondria) and tissues (Ref. Reference Ren, Sowers and Zhang163) may be the key to continuously respond to metabolic stress and will be crucial to preserving local and whole-body functions throughout the lifespan. Recent findings suggest that autophagy-deficient immune cells display features of premature aging, inflammation, high levels of ROS, mitochondrial dysfunction and a higher rate of cell death (Refs Reference Liu9, Reference Kaushik46, Reference Hansen, Rubinsztein and Walker164). Further, CD4+ T cells with lower rates of autophagic genes experience higher impairments in proper differentiation. Consequently, these impaired differentiated T cells will respond poorly to antigens, with abnormal secretions of inflammatory cytokines and higher rates of apoptotic genes, when compared to normal T cells.
Exercise training improves cellular autophagy although the exact mechanism is still elusive, as we and others summarised (Ref. Reference Batatinha165). Therefore, continuing to investigate the effective exercise training regimen, intensity and duration as well as the molecular events responsible for exercise-, age- and obesity- related responses in naïve CD4+ T cells is crucial to find therapeutic strategies to combat local and systemic inflammaging. More research is needed also to fine-tune the interaction of these endogenous and environmental factors to adequately prescribe the therapeutic strategy (pharmacologic or non-pharmacologic) in older individuals with metabolic comorbidities such as diabetes, non-alcoholic liver disease and metabolic syndrome. The scarce evidence presented in this review suggests that some metabolic dysfunctions may suppress autophagic benefits to exercise stimulus. Thus, it is important to determine the underlying mechanisms related to this pathology suppression to improve adaptive immune functions and autophagy abilities in these subjects as well as find adjuvant or alternative treatments.
Topical summary
Age, physical fitness and body composition have a great impact on autophagic (i.e., JNK1-2, ULK1,-2, Parkin/Pink-1, LC3B, Hsp70) and apoptotic gene expression (i.e., caspase3, Bim) as well as on differentiation of naïve CD4+ T cells and their interleukin secretions. To date, evidence between these endogenous and environmental factors is scarce among aged and young individuals. There is also a knowledge gap on the role of different physical fitness levels (sedentary and trained) and exercise training prescription modes (type, intensity, duration and volume) on autophagy response and naïve CD4+ T cells immunological functions. In this review we summarise the current state of knowledge of autophagy, with an emphasis on naïve CD4+ T cells and its modulation by endogenous and environmental cues namely aging, obesity and exercise.
Acknowledgement
We thank the São Paulo Research Foundation (FAPESP) and Coordenação de Aperfeiçoamento Pessoal de Nível Superior – Brazil (CAPES) #001. CSP is granted with Post Doctorate scholarship from the FAPESP (Process 2018/23402-0) and FSL is granted with research grants from the FAPESP (Process 2018/19678-0).
Conflict of interest
The authors declare that there are no conflicts of interest and all authors approved the final version before submission.



